摘要 针对芥子气(HD)染毒血浆的溯源性分析需求,采用超高效液相色谱串联质谱技术(UHPLCMS/MS),建立了人血漿中6种HD生物标识物(TDG,TDGO,SMO,SBMSE,MSMTESE,SBSNAE)的高灵敏度和专属性检测方法。应用甲醇和乙腈混合有机溶剂沉淀HD染毒血浆中的蛋白,然后用HLB柱固相萃取(SPE)纯化,经超高效液相色谱梯度洗脱分离,在三重四极杆串联质谱正离子多反应监测(MRM)模式下进行定性与定量检测。结果表明,6种目标物的线性范围为0.05~500ng/mL(R2=0.9840~0.9955),检测限在0.01~1.0ng/mL之间,各分析物的精密度(n=5)≤5.5%,加标回收率在86.5%~110.8%之间。本方法通过SPE的引入和UHPLC程序的优化,有效地解决了基质背景对分析可靠性的影响问题,并成功应用于国际禁止化学武器组织(OPCW)第5次生物医学样品分析演练HD染毒血浆样品的鉴定。
关键词超高效液相色谱串联质谱;芥子气;生物标识物;血浆
1引言
芥子气(HD)是一种糜烂性化学毒剂,化学名为2,2′二氯二乙硫醚,可通过吸入、皮肤和眼睛暴露等方式中毒,造成皮肤、眼睛、呼吸道等出现不同程度的损伤[1,2\]。两次世界大战以及80年代爆发的两伊战争中都使用了大量HD,造成了大量军民伤亡[3~5\]。侵华日军战败后在中国境内遗弃了大量包括芥子气在内的化学毒剂,长期以来对环境以及附近平民都造成了严重危害[6,7\]。1997年OPCW发布并生效的《化学武器公约》(CWC)严格禁止了HD的生产、储存和使用,但至今其销毁工作还未完成;同时由于HD容易生产、性质稳定,一旦被不法分子利用,还会极大地威胁公共安全。因此,有必要发展并建立灵敏、可靠的分析方法对HD染毒样品进行准确鉴定,对违约使用的调查取证、染毒人员的诊断救治都具有重要意义[8\]。
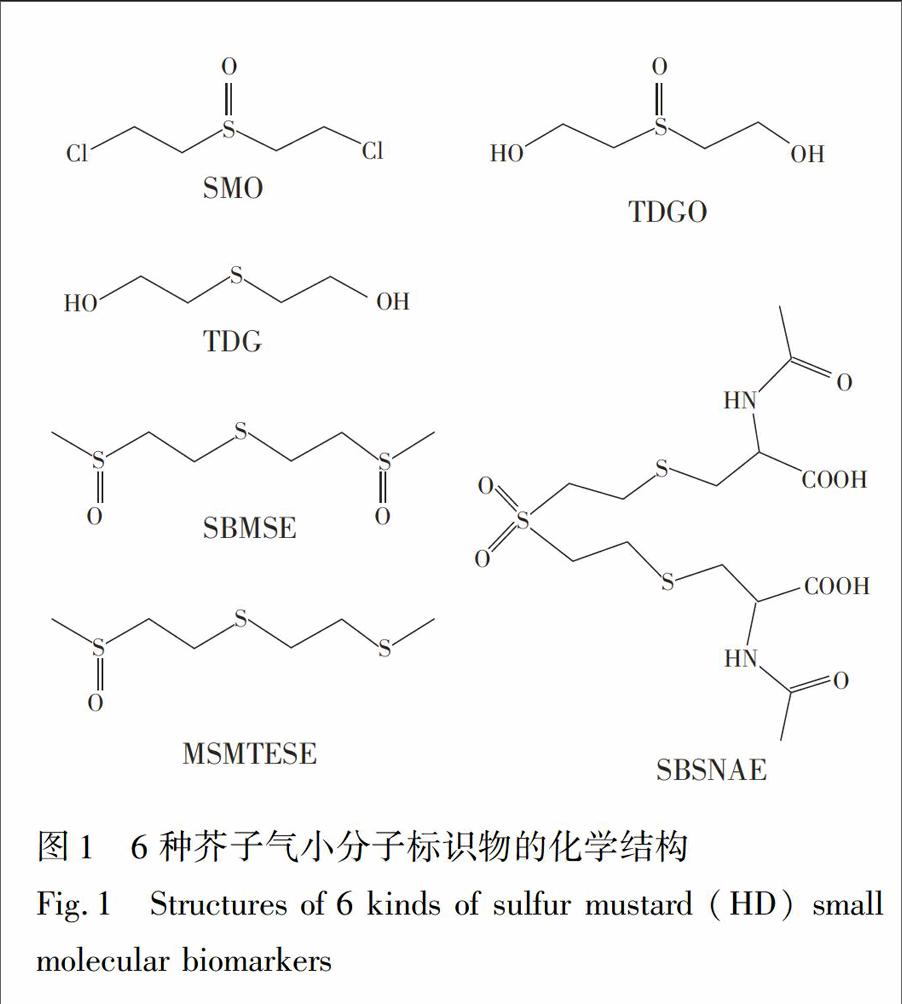
HD含有两个亲电中心,进入生物体后迅速分布于各组织中,发生代谢反应可能生成水解氧化产物、β裂解产物和大分子加合物等一系列化合物[9,10\]。这些代谢物按照其来源及特点分为两类:小分子游离代谢物和大分子加合代谢物[11~13\]。小分子游离代谢物包括硫二甘醇(TDG)、硫二甘醇亚砜(TDGO)及芥子气亚砜(SMO)等水解、氧化产物,以及与谷胱甘肽加合后经β裂解酶作用产生的1,1′磺酰基双\[2(甲基亚磺酰基)乙烷\](SBMSE),1甲基亚磺酰基2\[2(甲硫基)乙基磺酰基\]乙烷(MSMTESE),1,1′磺酰基双\[2S(N乙酰半胱氨酰基)乙烷\](SBSNAE)等β裂解产物(如图1所示)。在小分子代谢物中,虽然文献\[5\]报道了在生物体内检测出痕量的内源性TDG和TDGO,但在HD染毒的生物医学样品中,TDG与TDGO的浓度显著升高,而且这些化合物均具有HD的结构特征,因此,这些小分子代谢物均可作为HD生物标识物[11\]。与大分子加合物相比,小分子标识物在芥子气暴露后很快出现在体液中,能够在体内存在数日至两周,并且具有检测快速的优点,是染毒早期快速诊断的理想标识物。
对于HD小分子标识物的分析,大多采用GCMS/MS或LCMS/MS等技术分析其衍生产物或原体[14~17\],但由于不同标识物的性质差异,这些报道大都只针对单个或两个标识物进行分析。但HD染毒剂量、方式以及染毒时间的不同所产生的生物标识物也不尽相同[16\],因此,所建立的方法应能更广泛用于标识物和样品的分析。直到2013年,Li等[18\]报道了将血浆蛋白沉淀后直接进行LCMS/MS的分析方法,可同时检测7种HD游离代谢物。方法制备简单、耗时短,取得了较好的结果。但该方法中血浆经蛋白经沉淀直接分析,基质依然复杂,背景中存在大量干扰峰,分析大量样品后会造成色谱柱压升高、响应降低、仪器稳定性变差等问题。
针对以上情况,本研究引入SPE制备方法,并优化了UHPLC的梯度洗脱程序,建立了血浆中6种HD标识物(TDGO,SBMSE,SBSNAE,TDG,MSMTESE和SMO)的UHPLCMS/MS定量分析方法。实验结果表明,本方灵敏度高、选择性好、样品基质纯净,大批量样品分析响应稳定、重复性好。JP
2实验部分
2.1仪器、材料及试剂
1290超高效液相色谱系统(美国Agilent公司);6460型三重四极杆串联质谱系统(美国Agilent公司);Centrifuge5424型高速离心机(德国Eppendorf公司);NEVAP111型氮气吹干仪(美国OrganomationAssociates公司)。
HD、TDG、TDGO、SMO、SBMSE、MSMTESE及SBSNAE(均由本单位合成,NMR分析纯度>95%);HLB固相萃取柱(500mg/6mL,美国Waters公司);水、甲醇、乙腈(色谱纯,美国Honeywell公司);甲酸铵(美国RoeScientific公司);健康人血浆部分由北京红十字中心提供,部分来自于本实验室的自愿者。JP
2.2安全措施
所有涉及到芥子气的操作,如取用、配制和染毒过程均需要佩戴相应的防护器材,并在通风良好的状况下进行;以NaOH乙醇(1KG-3∶KG-510~1KG-3∶KG-511,V/V)混合溶液作为洗消剂,各种直接接触芥子气的实验器材在使用后立即洗消。
2.3染毒及加标血浆的制备
用乙腈配制1.0mg/mLHD标准工作液,吸取4.0μL标准工作液,加入4.0mL空白血浆,混匀、37℃振荡反应12h,得到1.0μg/mLHD染毒血浆储备液。用纯水配制6种HD标识物的混合标准液,各标识物浓度均为100μg/mL,用空白血浆稀释为0.01~500ng/mL的加标血浆。以上储备液均于
20℃保存。
2.4样品制备
参照文献\[18\]方法对血浆样品进行蛋白沉淀。0.5mL血浆中加入2.0mL乙腈甲醇(4KG-3∶KG-51,V/V),涡旋混匀,6100r/min离心5min,移取上清液。向沉淀中继续加入1.0mL乙腈甲醇(4KG-3∶KG-51,V/V),涡旋混匀、12200r/min离心5min,将两次的上清液合并,氮气吹干,1.0mL纯水复溶,离心收集上清液,加入60μL1.4%氨水准备进行SPE。
先后用5mL甲醇、12mL水和5mL1.4%氨水平衡HLB柱;上样,分别用3mL1.4%氨水和2mL2%甲醇1.4%氨水淋洗;最后用4mL含2%甲酸甲醇进行洗脱;浓缩洗脱液至近干,加入0.15mL纯水复溶,离心后取上清液进行LCMS分析。
2.5液相色谱质谱分析条件
色谱条件:ZorbaxEclipsePlusC18色谱柱(100mm×2.1mm,1.8μm,美国Agilent公司);流动相A为5mmol/L甲酸铵溶液,B为含5mmol/L甲酸铵的甲醇溶液;洗脱梯度:0~2.5min,5%~10%B;2.5~5.0min,10%~100%B;5.0~8.0min,100%B;8.0~8.1min,100%~5%B,运行3min;流速0.3mL/min;柱温40℃;进样体积10μL。
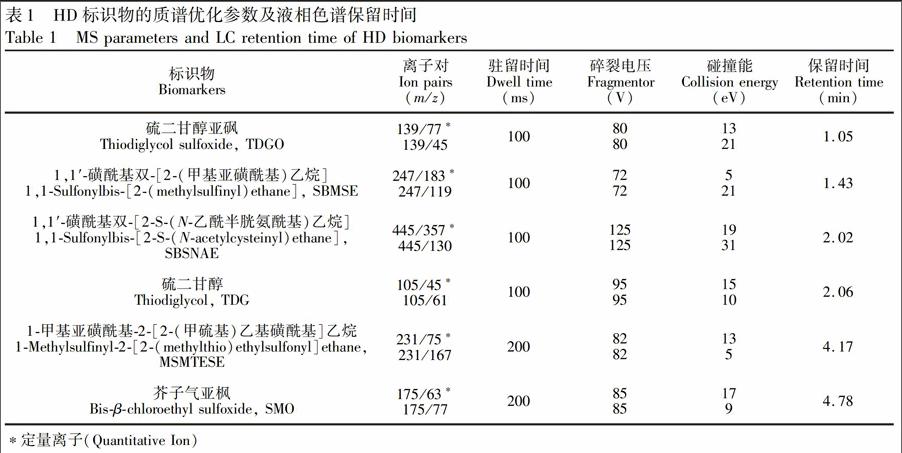
质谱条件:在ESI+模式下采用MRM方式扫描,应用4个时间段扫描6个目标物的定量及定性离子对,0~1.75min扫描SBSME和TDGO;1.75~3.60min扫描SBSNAE和TDG;3.60~4.65min内扫描MSMTESE;4.65min后扫描SMO。毛细管电压4.0kV,离子源温度350℃;雾化气压力35psi;碰撞加速电压3V,权重为1/x。其它参数见表1。
3结果与讨论
3.1固相萃取条件的选择
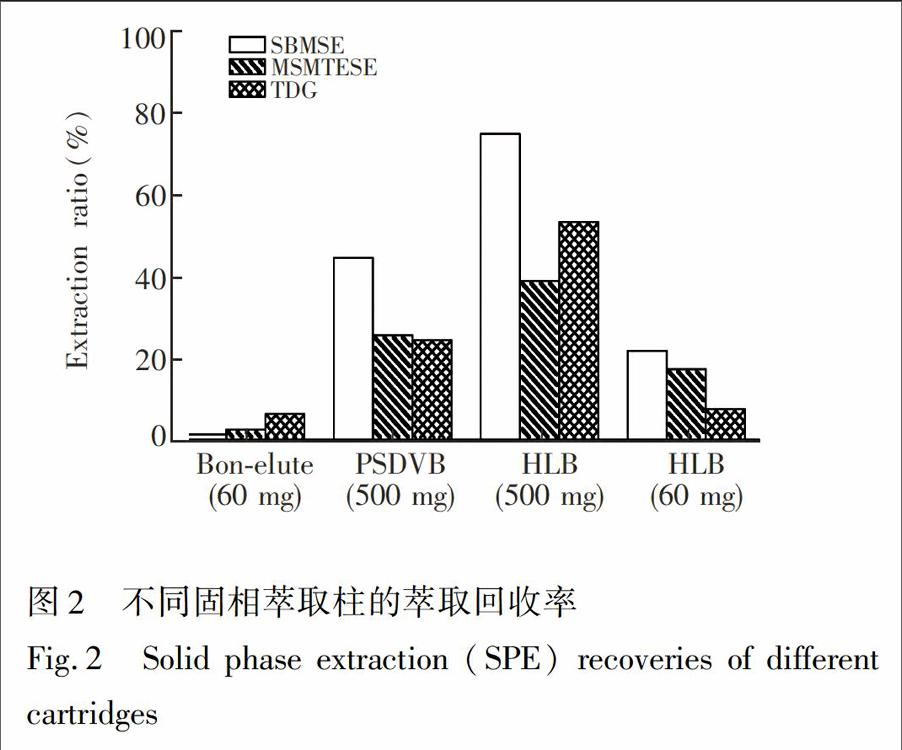
与文献\[18\]相比,针对血浆基质的复杂性,增加了固相萃取步骤,目标是进一步除去基质中的背景干扰物,使制备后的样品更加纯净,提高检测灵敏度和定量稳定性。6种化合物的极性顺序为TDGO>SBMSE>SBSNAE>TDG>MSMTESE>SMO,其中TDGO与SBMSE,SBSNAE与TDG,MSMTESE与SMO的极性相近,因此,选择3种化合物SBMSE、TDG和MSMTESE进行血浆加标(浓度均为10ng/mL),考察了BondElutePlexa(60mg,3mL),SampliQPSDVB(500mg,6mL),HLB(500mg,6mL),以及HLB(60mg,3mL)JP4种SPE柱的富集效果,结果见图2。
BondElutePlexa柱的填料为疏水性的二乙烯及苯基聚合物,主要用于富集弱极性化合物,而SBMSE及TDG等目标物的极性相对较强,因此回收率低。而PSDVB柱的填料为聚苯乙烯二乙烯基苯共聚物,适用于从水溶液中提取极性芳香化合物,如苯酚等,不适于本文目标物的富集。HLB采用二乙烯苯和N乙烯基吡咯烷酮的亲水亲脂两性填料,可富集的化合物极性范围更宽。此外,还考察了60mg小柱床的HLB柱,以期在不损失回收率的情况下,减少背景干扰,但实验结果表明60mg的SPE柱的容量有限,回收率偏低,不适于0.5mL血浆样品的富集。因此,经比较,本实验最终选择回收率最高的HLB(500mg/6mL)柱对6种HD标识物的加标样品进行SPE纯化。
目标化合物都显弱碱性,加入60μL1.4%氨水,目的是降低目标化合物的极性,增强与HLB柱填料疏水基团的结合。经过比较考察,最终选择先用3.0mL1.4%氨水淋洗残留在柱上的杂质,再用2.0mL含1.4%氨水的2%甲醇淋洗柱子上非特异性结合的极性干扰物。此外,向洗脱液中添加2%甲酸是为了降低目标物在HLB柱上的结合力,使其更容易被洗脱。实验结果表明,SPE在有效富集目标物的同时,除去了血浆基质中的大量背景干扰,色谱图基线干净(图3),延长了色谱柱的使用寿命。
3.2UHPLCMS/MS方法的优化
根据6种化合物的理化性质及三重四极杆质谱的特点,在ESI+模式下应用Masshunteroptimizer軟件对每一个目标化合物的分析参数进行优化,结果见表1。为了在较短时间内获得良好的分离效果,对洗脱程序进行优化。由于TDGO和SBMSE这两种化合物极性很强,所以在前2.5min内没有采用等度平衡程序,目的是将目标物与基质中的强极性背景化合物进行分离,减少干扰;MSMTESE和SMO在色谱柱上保留较好,为了缩短分析时间,采用快速梯度洗脱程序对目标物进行洗脱。在优化的洗脱条件下,6种HD标识物在6min内均有较好的分离(图3),TDG和SBSNAE的保留时间重合,可通过MRM离子对m/z的差异进行区分。此外,本研究采用4个时间段同时分析6个HD代谢物的12个MRM离子对,使每个峰能获得定量分析所需的足够扫描点数,改善峰形和灵敏度。
3.3方法学考察
3.3.1专属性在最佳色谱质谱条件下,测试溶剂空白(纯水)、空白血浆、加标血浆(6种标识物浓度均为50ng/mL)和HD染毒血浆(100ng/mL)样品。溶剂空白中未检出任何相关化合物(图4A),表明仪器系统中没有相关背景干扰。空白血浆中仅检测到少量内源性TDGO和TDG(图4B),浓度分别为2~8ng/mL和0~5ng/mL,与文献[5]报道一致。50ng/mL加标血浆样品中6种化合物的响应良好(图5A)。在100ng/mLHD染毒血浆中发现了显著高于空白血浆的TDGO和TDG,可以作为HD染毒依据(图5B)。在体外染毒血浆样品中没有检测到SMO的原因是在低浓度染毒时HD主要发生水解反应,自身氧化反应很少[8\]。β裂解产物是由HD与谷胱甘肽的加合物在β裂解酶的作用下而生成,但谷胱甘肽主要存在于血细胞中,离心后的血浆中谷胱甘肽含量极少,所以未检测到。但是,在真实染毒患者体内,HD既可能发生水解、氧化反应,还可能与血液中的谷胱甘肽充分反应后释放出β裂解产物,染毒人员血液经离心得到的血浆中可能存在水解氧化及β裂解标识物[18\]。因此,本方法依然可应用于真实HD染毒患者体内代谢后的血浆样品分析。4种样品的MRM色谱图比较结果显示,空白血浆中除低水平的TDGO和TDG内源性本底外,无其它干扰物,表明本方法专属性较好。
3.3.2线性范围、方法检出限及定量限为了降低基质效应对检测准确度的影响,本研究采用基质标准曲线外标法进行定量分析。将浓度为0.01~500ng/mL的加标血浆按照2.4和2.5节进行处理分析。以目标物的浓度和峰面积进行线性回归,权重为1/x,绘制定量标准曲线。以响应显著高于空白血浆且信噪比为3对应的浓度作为方法的检出限(LOD),信噪比为5且相对标准偏差(RSD)低于20%所对应的浓度作为方法的定量限(LOQ)[18\],结果见表2。6种目标物均有较宽的线性范围,SBMSE、SBSNAE和MSMTESE的检出限与文献\[18\]报道一致,而SMO、TDG与TDGO的灵敏度比文献值提高了5~10倍(表3)。
3.3.3加标回收率与精密度对5,150和450ng/mL的加标血浆质量控制样品(LQC,MQC和HQC)进行平行处理并分析,每个样品重复测定5次。计算各目标化合物的精密度和回收率,结果见表4。6种化合物的回收率在86.5%以上,RSD<5.5%,均符合方法学要求。
3.4HD染毒样品分析
CM(42OPCW在2015年第5次生物医学样品分析演练中提供了6份怀疑HD中毒人员的血浆样品CM)LM
(A~F),和1份空白血浆(M)。采用所建方法对所ZH(有
样品进行定性及定量分析,检测结果见图6。在C样品中检测到的TDG及TDGO与血浆基质M一致,所以确定C样品为空白血浆;在A,B,D,E和F样品中都检测到高于内源水平的TDG与TDGO,根据检测得到TDG与TDGO的含量大小,得出样品染毒浓度顺序为F>A>E>D>B。OPCW发布的结果中C为空白血浆,A,B,D,E和F分别为100,10,40,50和150ng/mLHD染毒血浆,说明本方法的CM(21*3/4检测结果与OPCW官方提供的染毒信息一致。空CM)ZH)
白血浆中内源TDG与TDGO的分析结果表明,HD对血浆的染毒浓度在10ng/mL以上时,可利用TDG和TDGO作为标识物进行HD的溯源性分析,而之前未有此方面报道。另外有意义的是,本方法检测到的TDG与TDGO的浓度和与HD染毒浓度(10~150ng/mL,OPCW提供)呈良好的线性关系,这使得依据检测染毒血浆样品中标识物的浓度检测实际HD染毒浓度成为可能,但由于代谢物的浓度还与染毒时间密切相关,特别是在体内染毒情况下,因此,代谢标识物与染毒浓度之间的关系还需要进行深入探讨。
References
1(#GhabiliK,AgutterPS,GhaneiM,AnsarinK,PanahiY,ShojaMM.Crit.RevToxicol.,2011,(41):384-403
2KeheK,SziniczL.Toxicology,2005,(214):198-209
3KeheK,ThiermannH,BalszuweitF,EyerF,SteinritzD,ZilkerT.Toxicology,2009,263(1):3-8
4LawrenceRJ,SmithJR,BoydBL,CapacioBR.J.Anal.Toxicol.,2008,(32):31-36
5ZhangY,YueL,NieZ,ChenJ,GuoL,WuB,FengJ,LiuQ,XieJ.J.Chromatogr.B,2014,(961):29-35
6HuaX,ZhiYN,ZhangYJ,LiCZ,YueLJ,YangWF,ChenJ,DongY,LiQ,LinY,WuBD,FengJL,LiH,GuoL,XieJW.Toxicol.Rep.,2014,(1):533-543
7DavisKG,AsperaG.Ann.Emerg.Med.,2001,(37):653-656
8RuffAL,DillmanJF.Eplasty,2007,(8):e2
9AndachtTM,PantazidesBG,CrowBS,FidderA,NoortD,ThomasJD,BlakeTA,JohnsonRC.J.Anal.Toxicol,2014,(38):8-15
10PantazidesBG,CrowBS,GartonJW,QuionesGonzlezJA,BlakeTA,ThomasJD,JohnsonRC.Chem.Res.Toxicol.,2015,28(2):256-261
11NieZ,LiuQ,XieJ.Talanta,2011,85(2):1154-1159
12LinY,DongY,ChenJ,LiCZ,NieZY,GuoL,LiuQ,XieJW.J.Chromatogr.B,2014,,15(945946):233-239
13WeiY,YueL,LiuQ,ChenJ,XieJ.J.Chromatogr.B,2011,(879):1707-1712
14RichesJ,ReadRW,BlackRM.J.Chromatogr.B,2007,(845):114-120
15LIChunZheng,CHENJia,ZHONGYuHuan,ZHONGYuXu,XIEJianWei,LIHua.ChineseJ.Anal.Chem.,2012,40(10):1567-1572
李春正,陳佳,钟玉环,钟玉绪,谢剑炜,李桦.HTK分析化学,2012,40(10):1567-1572
16BoyerAE,AshD,BarrDB,YoungCL,DriskellWJ,WhiteheadRDJr,OspinaM,PrestonKE,WoolfittAR,MartinezRA,SilksLA,BarrJR.J.Anal.Toxicol.,2004,28(5):327-332
17NIEZhiYong,ZHANGYaJiao,WUBiDong,YANLong,FENGJianLin,LIUQin,XIEJianWei.ChineseJ.Anal.Chem.,2014,42(7):980-984
聶志勇,张雅姣,吴弼东,闫珑,冯建林,刘勤,谢剑炜.HTK分析化学,2014,42(7):980-984
18LiC,ChenJ,LiuQ,XieJ,LiH.J.Chromatogr.B,2013,917918:100-107)
、
AbstractAsensitivemethodwasdevelopedforthesimulationdeterminationofsixkindsofsulfurmustards(HD)biomarkers(TDGO,SBMSE,SBSNAE,TDG,MSMTESEandSMO)inhumanplasmabyultrahighperformanceliquidchromatographytandemmassspectrometry(UHPLCMS/MS).HDexposedplasmasampleswerepretreatedwithamixedsolventofmethanolandacetonitriletoprecipitateproteins.Asolidphaseextraction(SPE)methodwithHLBcartridgewasusedtopurifythetargetbiomarkersfromthematrix.AcompleteseparationwasachievedonanUHPLCsystemwithagradientelution.Thequalitativeandquantitativeanalyseswerecarriedoutbytriplequadrupoletandemmassspectrometryinmultiplereactionmonitoring(MRM)mode.Theresultsshowedthatthecalibrationcurvesforthe6biomarkerswerelinear(R2=0.9840-0.9955)overtherangefrom0.05to500ng/mL,withthelimitsofdetection(LODs)of0.01-1.0ng/mL.Therelativestandarddeviation(RSD,n=5)was≤5.5%,andtherecoveryoftheanalytesrangedfrom86.5%to110.8%.Theinfluenceofthematrixbackgroundsonthemethod'srobustnesswaseffectivelydiminishedbyintroducingSPEprocedureandbyoptimizingtheUHPLCelutionprogram.ThemethodwassuccessfullyappliedtothefifthbiomedicalsampleanalysisexerciseorganizedbytheOrganizationfortheProhibitionofChemicalWeapons(OPCW).
KeywordsUltrahighperformanceliquidchromatographytandemmassspectrometry;Sulfurmustard;Biomarkers;Plasma
HQWT6JY(Received8May2016;accepted23August2016)
- 情境教学在高中政治教学中的应用
- 时事政治在高中政治教学中的应用
- 浅析核心素养背景下的初中数学深度学习策略
- 浅析初中数学教学引导学生自主学习策略
- 多种方式开展初中历史活动课
- 课外阅读在小学语文作文教学中的必要性探究
- 小学音乐唱歌教学中存在的问题及对策
- 重视小学语文阅读教学,提高小学语文教学效率
- 农村初中数学阅读能力培养的策略研究
- 刍议高中政治中趣味教学法的应用
- 小学体育兴趣化教学在田径教学中实践运用
- 初中数学课堂师生互动策略探讨
- 小组合作学习在高中英语教学中的应用
- 高中生物教学中德育渗透措施探究
- 初中化学教科书中探究活动的设计分析
- 核心素养视角下的初中物理高效课堂的有效构建
- 农机技术与农艺技术深度融合初探
- 幼儿园安全管理工作优化策略浅谈
- 浅析小学生英语语感的培养对策
- 初中英语课堂教学存在的问题及优化策略
- 如何在初中历史教学中进行创新教学
- 浅谈案例教学法在初中政治教学中的应用
- 关于初中物理高效课堂的探索与实践
- 立德树人理念下的小学班主任德育工作策略探究
- 高中英语教学中思维导图的应用
- litres
- lits
- litter
- litter bin
- littered
- littering
- litters
- litter²
- litter¹
- little
- little boy/little girl
- little-by-little
- little by little/bit by bit
- little chance/hope/possibility/prospect
- little did i/she/he etc know
- little known
- little-known
- little known/little-known
- littleness'
- littleness
- littlenesses
- littleness's
- littler
- littles'
- littles
- 神采奕然
- 神采才华
- 神采焕发
- 神采焕然
- 神采艳丽
- 神采英发
- 神采英拔
- 神采风度
- 神采飞扬
- 神采飞扬,精神振奋
- 神鉴
- 神钲
- 神钲迢递于高峦
- 神锋
- 神镜高悬
- 神门
- 神闲意定
- 神闲气定
- 神闲气静
- 神雀五凤甘露黄龙之瑞
- 神雨
- 神霄绛阙
- 神靈
- 神鞭
- 神韵