
摘 要 脱氧核酶(DNAzyme或DNA酶)是一类具有高效催化活性和特异性识别功能的DNA分子,可以通过体外筛选方式从随机脱氧核苷酸单链库中获得,它具有催化效率高、特异性高、稳定性好、合成简单且修饰方便等优点。脱氧核酶与纳米材料的结合, 既保留了脱氧核酶的催化特性和识别能力, 又引入了纳米材料的信号转导功能,实现了识别与信号转导功能的一体化,大大促进了生物传感器的快速发展。本文主要介绍了金纳米颗粒、石墨烯、量子点、磁性纳米颗粒等纳米材料结合脱氧核酶用于生物传感的研究进展。
关键词 脱氧核酶; 纳米材料; 生物传感器; 评述
1 引 言
纳米材料是由粒径小于100 nm的超细颗粒构成的零维、一维、二维、三维材料的总称。由于纳米材料具有独特的尺寸结构,所以有着传统材料不具备的一些特征[1]:(1)表面效应 指随着微粒粒径的变小, 其表面原子数与总原子数之比急剧增大, 从而引起纳米微粒性质的变化。由于这些纳米微粒表面原子处于严重的缺位状态, 使得它易与其它原子结合, 达到稳定状态,故具有很高的化学活性。(2)小尺寸效应 指由于纳米颗粒的尺寸变小所引起的光、力、热、电、磁等宏观物理性质的变化。如纳米颗粒的熔点远低于块状金属的熔点; 当金属纳米颗粒的直径小于10 nm时,就会失去原有的金属光泽,呈现出黑色。(3)量子尺寸效应 指当颗粒的尺寸下降到纳米级时,费密能级附近的电子能级就会由准连续态变为分立能级,能级间的间距随颗粒尺寸的减小而增大。当热能、磁场能或电场能比平均的能级间距还小时,纳米微粒就会呈显出一些与宏观物体截然不同的特性,如由导电的金属制备的纳米颗粒可以变为绝缘体。(4)宏观量子隧道效应 是指微观粒子贯穿势垒的能力。纳米材料的这些特征使其表现出一系列独特的光学、电学、力学、磁学以及催化性能[2,3]。目前, 纳米材料已经被广泛应用于信息工程、能量储存、生物传感、分子器件及选择性催化等研究领域, 并逐渐成为科研人员关注的焦点[4~7]。
脱氧核酶(DNAzyme或DNA酶)是一类具有高效催化活性和特异性识别功能的DNA分子,可以通过体外筛选方式从随机脱氧核苷酸单链库中获得[8~12]。脱氧核酶可以催化DNA或RNA切割、DNA水解以及DNA连接等多类反应[7,12~15],但常见的脱氧核酶主要包括RNA切割型脱氧核酶与G四聚体脱氧核酶两大类。与传统蛋白酶相比,脱氧核酶具有以下优点: 首先,它的稳定性高,在较高温度下其活性不受影响;其次,脱氧核酶的相对分子量比较小且具有很高的催化效率; 此外,脱氧核酶的合成简单、修饰方便并且受酸度等环境因素影响比较小。脱氧核酶的上述优点使其受到了越来越多研究者的关注,并且已被广泛应用于多个研究领域[16~20]。如脱氧核酶可以作为一种工具酶,用于细胞水平上的基因敲除,即特异性地使某一基因失活, 然后观察该基因在细胞生理、生化中的作用,研究该基因的功能,以便用于抗病毒、抗肿瘤的基因治疗[19,20]。由于具有高效催化活性和对辅因子依赖性的特异性识别功能,脱氧核酶也可用于生物传感器的构建[21~23],但如何将脱氧核酶与目标物的识别信息转化为可检测的物理信号,一直是生物传感研究领域的难点和热点。
脱氧核酶与纳米材料的结合既保留了脱氧核酶的催化特性和识别能力, 又引入了纳米材料的信号转导功能,实现了识别和信号转导功能的一体化,有利于设计出高灵敏度、高选择性及高效的生物传感体系,从而为分析化学和材料科学的发展提供了新动力。基于上述优点,脱氧核酶已与石墨烯、纳米金、量子点和磁性纳米颗粒等纳米材料广泛结合,发展了一系列新型生物传感器。本文将围绕各种新型纳米材料在脱氧核酶生物传感器中的应用进行评述。
2 脱氧核酶-纳米金复合物在生物传感中的应用
纳米金颗粒(Gold nanoparticles, AuNPs)又称胶体金或金溶胶,具有大的比表面积、高催化活性以及尺寸依赖的光学性质等独特的物理化学性质[24]。纳米金颗粒的制备技术与表面修饰技术的发展提高了它的稳定性、水溶性和生物相容性,拓宽了其在生物分子标记和检测、纳米生物传感器以及纳米生物芯片等方面的应用[25~27]。
2.1 基于脱氧核酶-纳米金颗粒的比色型传感器的研究
纳米金颗粒是在比色传感器中常用的一种信号探针,其表面等离子体效应与自身粒径的大小以及颗粒之间的距离有关。当纳米金颗粒由分散状态变为团聚状态时,其颜色会由红色变为蓝色或紫色。基于上述原理, 科研工作者发展了一系列比色型的脱氧核酶传感器[28~33]。
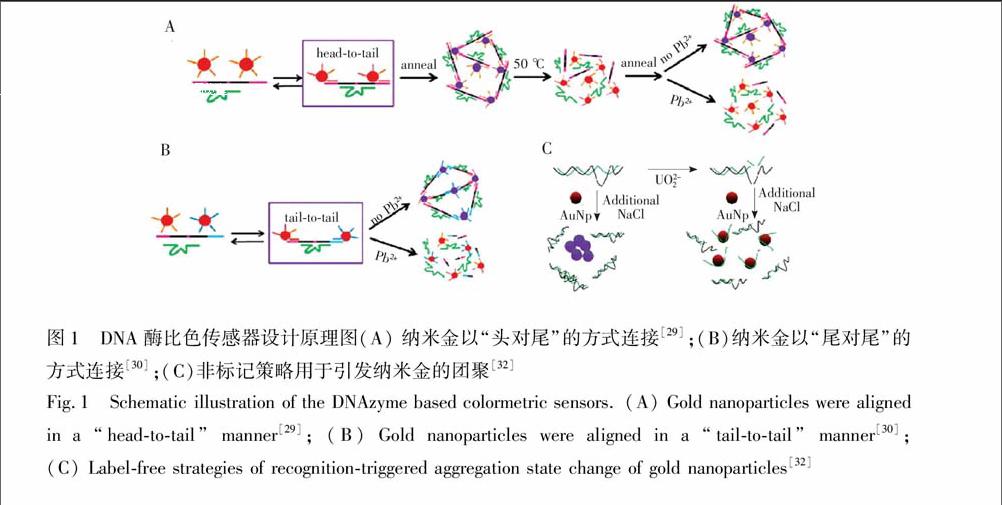
Lu研究组[29]首次将DNAzyme通过SAu键共价修饰于纳米金表面构建了一种可用于检测Pb2+的比色传感器。如图1A所示,底物链的两端延长, 便于与金纳米颗粒表面修饰的DNA以头对尾的方式杂交。当不存在Pb2+时,底物链与酶链以及纳米金表面修饰的DNA之间形成稳定杂交,纳米金颗粒团聚,溶液显示蓝色。当加入Pb2+后,底物链被切断, 抑制了纳米金的团聚,溶液显红色。该传感器的检出限为100 nmol/L,可用于油漆中Pb2+的检测。然而该传感器采用头对尾的杂交方式会产生很大的空间位阻,因此需要复杂的加热-冷却过程。为了解决这个问题,该小组又采用了尾对尾的杂交方式, 降低了杂交产生的空间位阻, 并使反应可以在室温下进行[30](图1B)。此外,他们又引入了一段可与切割后的底物链片段杂交的DNA序列, 用于促进纳米金的释放,使得传感器的响应时间只需5 min[30]。由于上述DNA酶传感器都需要将DNAzyme共价修饰在纳米金表面,过程复杂、成本较高, 且耗时长,因此该小组又基于DNA酶切割底物链可产生单链DNA,而单链DNA能保护纳米金颗粒在较高盐离子条件下不会团聚,设计了一种非标记的比色传感器用于Pb2+和UO2+2的检测[32](图1C)。与此同时,Wang研究组[33]也报道了类似的无标记检测Pb2+的比色型DNA酶传感器。除此之外,Lu研究组[34]还基于脱氧核酶的催化连接活性, 设计了一种比色型传感器, 用于Cu2+的检测,该传感器的背景干扰小,灵敏度高。上述结果表明,标记型传感器需要较长时间进行预处理和制备,但是当制备好传感器后更易
图1 DNA酶比色传感器设计原理图(A) 纳米金以“头对尾”的方式连接[29];(B)纳米金以“尾对尾”的方式连接[30];(C)非标记策略用于引发纳米金的团聚[32]
Fig.1 Schematic illustration of the DNAzyme based colormetric sensors. (A) Gold nanoparticles were aligned in a “head-to-tail” manner[29]; (B) Gold nanoparticles were aligned in a “tail-to-tail” manner[30]; (C) Label-free strategies of recognition-triggered aggregation state change of gold nanoparticles [32]于操作和检测;而非标记型传感器的灵敏度更高,且无需长时间制备、更加经济,然而在检测过程中易受到离子强度及其它一些因素的影响。
2.2 基于脱氧核酶-纳米金颗粒的荧光传感器的研究
纳米金颗粒除了可以作为信号报告基团用于比色分析外,还可用于荧光检测。人们利用纳米金颗粒作为荧光猝灭剂, 发展了一系列DNA酶荧光传感器[35~38]。Wang等[35]设计了一条底物链与酶链一体化的探针,并将该探针通过SAu键共
图2 A 基于纳米金作荧光猝灭剂的DNAzyme荧光传感器[36]; B 基于荧光偏振技术的DNA酶生物传感器原理示意图[38]
Fig.2 (A) Schematic illustration of DNAzyme-based fluorescent biosensors using gold nanoparticles as fluorescence superquencher[36] and (B) Schematic illustration of DNAzyme-based fluorescence anisotropy assay[38]
价修饰于纳米金颗粒表面。由于标记在底物链末端的荧光团会靠近纳米金,故荧光被猝灭。当加入Pb2+后, 底物链被切断,标记有荧光团的部分底物片段游离出来,荧光得以恢复,从而实现了Pb2+的定量检测(图2A)。此外,Malashikhina等[36]发展了一种DNA酶与纳米金结合的荧光生物传感器, 用于抗坏血酸的检测。Yin等[37]基于荧光偏振的方法设计了一种可以检测金属离子的DNA酶传感器(图2B)。标记有荧光团的底物链与共价修饰在纳米金上的酶链杂交,由于大分子在溶液中运动慢,故荧光各向异性值大。Pb2+存在时, 底物链被切割,标有荧光团的部分底物片段与纳米金分离,导致荧光各向异性值变小。然而, 上述传感器都仅限于体外生物分子的检测,构建可高灵敏和特异性检测体内生物分子的脱氧核酶生物传感器已成为近年来研究的热点。Lu课题组基于金纳米颗粒高的荧光猝灭率构建了一种可检测细胞内UO2+2的生物传感器[38]。DNA酶链共价修饰于纳米金表面,底物链两端分别标记有Cy3荧光团和BHQ猝灭团。当酶链与底物链杂交后,由于双重猝灭作用,Cy3的荧光被有效猝灭。当加入UO2+2后,UO2+2催化底物链的RNA水解,从而使标记有荧光团的部分底物片段游离出来,荧光得到恢复。该荧光纳米探针经内吞作用进入细胞后,可以实现活细胞内UO2+2的成像检测。
2.3 纳米金颗粒作为信号放大基团在生物分析中的应用
传统的检测方法中,探针的一端只能标记一个生物分子,
其灵敏度受到限制。而纳米金具有比较大的比表面积,作为探针载体其表面可标记多个生物分子,从而可以实现检测信号的放大[39~42]。
图3 基于树枝状纳米结构引发的放大传感策略用于Pb2+检测原理示意图[40]
Fig.3 Schematic of the amplified sensing strategy of DNA-Au dendrimer-based SERS biosensor for Pb2+ detection[40]
本研究组发展了一种基于树枝状纳米结构信号放大技术的新型SERS传感器[39](图3)。其底物链和酶链通过10个聚
T碱基连接在一起,且底物链的末端标记有巯基,可通过SAu键组装在金电极表面。当加入Pb2+后,底物链中RNA碱基被水解,使其被切割为两部分,与酶链连接的部分底物序列从金电极表面脱落下来,而金电极表面剩余的底物片段可与纳米金标记的报告探针杂交,然后通过层层自组装形成树枝状纳米结构,SERS信号得到显著增强,从而实现Pb2+的高灵敏检测。而Shen等[40]构建了一种以DNA功能化纳米金作为信号放大基团的DNA酶电化学传感器。标记有巯基的DNA酶链首先被组装在金电极表面,而报告DNA 探针标记在纳米金颗粒表面,底物链则分别与酶链和报告DNA探针杂交形成三明治结构。由于电活性物质六氨合钌可以嵌入报告DNA 探针中从而获得较大的电流信号。Pb2+的加入使得底物链被切断,导致标记有报告DNA 探针的纳米金颗粒从电极表面脱离,导致电化学信号减小,由此可以放大检测Pb2+,与不用纳米金放大基团相比,其灵敏度提高了5倍。
3 脱氧核酶-石墨烯复合物在生物传感中的应用
石墨烯(Graphene)是一种由碳原子通过sp2杂化构成的单层蜂窝状的平面薄膜,它可以折叠成零维的富勒烯,卷曲成一维的碳纳米管或堆垛形成三维的石墨,因此石墨烯是构成其它碳质材料的基本结构单元[43]。石墨烯具有比表面积大、机械强度高、导电性和导热性好且表面修饰方便等优点, 因而受到了很多科研工作者的关注。氧化石墨烯(GO)是石墨烯的衍生物,由于其表面含有很多羧基、羟基和环氧基等含氧活性基团,所以具有良好的水溶性和生物相容性。将石墨烯及其衍生物与脱氧核酶结合, 用于构建高效的生物传感器, 引起了科研人员的广泛关注[44~49]。
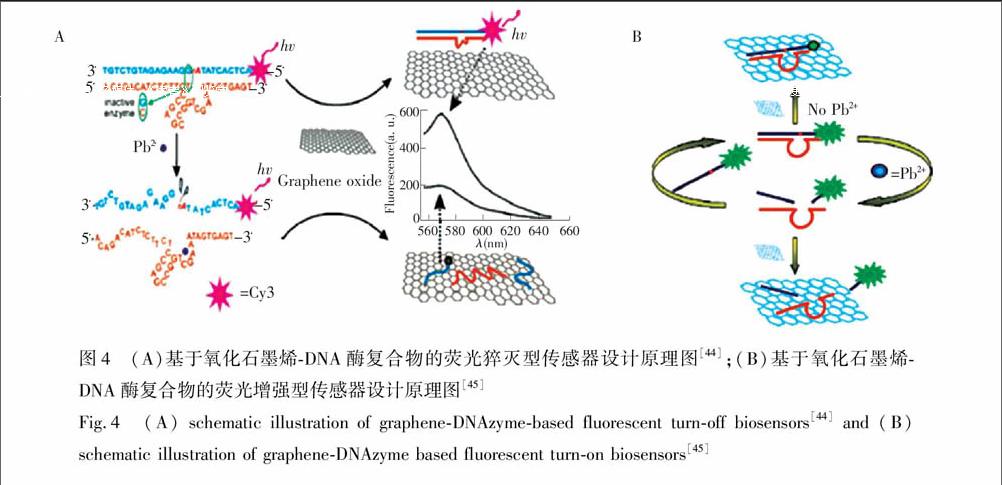
Wen等[44]利用氧化石墨烯对单双链DNA的吸附能力不同, 设计了一种DNA酶荧光探针, 用于Pb2+检测(图4A)。不存在Pb2+时,由于杂交探针的刚性双链结构使其与氧化石墨烯之间的吸附力比较弱,所以底物链上标记的荧光团的荧光不能被猝灭; 加入Pb2+后,底物链中RNA碱基被水解,使其被切割为两部分,此时标记有染料的部分底物片段与酶链解链并游离出来。由于游离的底物片段与氧化石墨烯之间吸附能力强,因此染料的荧光被猝灭,从而实现了Pb2+的定量检测。由于上述传感器是猝灭型的DNA酶传感器,其检测的灵敏度受到限制。为了提高传感器的灵敏度,本研究组[45]基于氧化石墨烯对不同长度的单链DNA具有不同的吸附能力, 设计了一种荧光增强型的DNA酶生物传感器, 用于Pb2+的高灵敏检测(图4B)。由于底物链和酶链的杂交探针中含有一段单链DNA序列,故该探针可以吸附在GO上,并使底物链5′端标记的荧光团靠近GO,导致荧光被猝灭。Pb2+的加入使DNA酶的催化活性被激活,底物链被切割为两段。标记有荧光团部分底物片段(5个碱基)与酶链解链。释放出的酶链可继续与未反应的底物链杂交,同时诱发下一轮反应。如此循环, 传感体系中就含有很多标记有荧光团的短链DNA。加入GO后,由于短链DNA与GO的结合力弱,故荧光团远离GO,其荧光不被猝灭。该传感器的灵敏度高,其检出限为300 pmol/L。此外,Yu等[46]基于氧化石墨烯可以增强荧光各向异性的策略发展了一种DNA酶传感器。上述传感器虽然灵敏度比较高,但是底物链都需要标记荧光团,故合成复杂且成本较高。基于此,Liu等[47]将核酸嵌入剂GelRed和氧化石墨烯结合构建了一种非标记的DNA酶荧光传感器, 用于Cu2+的检测。除了基于DNAzyme的催化作用检测其辅助因子外,DNAzyme
Fig.4 (A) schematic illustration of graphene-DNAzyme-based fluorescent turn-off biosensors[44] and (B) schematic illustration of graphene-DNAzyme based fluorescent turn-on biosensors[45] 在基因治疗中也显示出巨大的潜力。Kim等[48]利用氧化石墨烯作为载体将标记有荧光团FAM的DNA酶非共价吸附在石墨烯上, 并用于细胞内丙型肝炎病毒(HCV)基因的检测与沉默。
石墨烯除了可与RNA切割型脱氧核酶结合设计生物传感器外,也可与G四聚体脱氧核酶结合设计一些生物传感器[49~51],从而进一步拓宽可检测靶分子的类型,丰富传感器的设计模式。Luo等[50]将可识别目标序列的探针DNA与G-四聚体序列整合到一条链上,没有目标物存在时,探针DNA通过π-π键的相互作用吸附在GO上,从而使G四聚体-鲁米诺复合体靠近GO,导致化学发光信号很弱; 加入目标DNA后,目标DNA与探针DNA杂交形成双链刚性结构,从而远离GO, G四聚体-鲁米诺复合体的化学发光信号增强,由此可以定量检测目标DNA。为了进一步提高传感器的灵敏度,Yuan等[51]将功能化的石墨烯作为G四聚体-Hemin复合体的纳米载体, 设计了一个可以放大检测凝血酶的电化学传感器。
4 脱氧核酶-量子点复合物在生物传感中的应用
量子点(Quantum dots, QDs) 又称为半导体纳米晶,通常是由Ⅱ~Ⅵ或Ⅲ~Ⅴ族元素组成的新型半导体纳米材料, 直径在2~10 nm,由于电子和空穴被量子限域,其连续能带结构变成分立能级结构,故受激发后可以产生荧光。与传统有机染料相比,量子点具有激发谱带宽、发射波长可调、发光寿命长、荧光量子产率高及光化学稳定性好等优良的光学特性[53~56],并成为化学与生物传感领域中很有发展潜力的一种荧光纳米材料。目前,研究人员将脱氧核酶与量子点结合并在生物分析领域做了一系列工作[57~59]。Wu等[57]利用发光为530 nm和625 nm两种量子点构建了可同时检测Pb2+与Cu2+的DNA酶荧光传感器(图5A)。具体检测原理是标记有猝灭团的底物链共价修饰到量子点表面,而同样标记有猝灭团的DNA酶则与底物链杂交,由于量子点与猝灭团靠近, 发生荧光共振能量转移, 导致量子点的荧光被猝灭。当加入目标物后,DNA酶的催化活性被激活, 并将底物链上RNA碱基水解,使其被切割为两部分,故猝灭团远离量子点,量子点的荧光恢复,实现了Pb2+与Cu2+的同时检测。与传统的荧光染料相比,灵敏度分别提高了50倍和70倍。Sharon等[58]则基于G-四聚体脱氧核酶与血红素结合后可以通过电荷转移猝灭CdSe/ZnS量子点荧光, 设计了一种可以检测DNA和腺苷的荧光传感器(图5B)。
图5 基于脱氧核酶与量子点结合的生物传感器检测原理图[57,58,60]
Fig.5 Schematic illustration of DNAzyme-quantum dots-based biosensors[57,58,60]
除了可以利用量子点的荧光性质设计生物传感器外, 也可以利用其电化学性质检测多种目标物[60~62]。 Zhang等[60]利用标记有链霉亲和素的PbS量子点与DNA酶结合, 构建了一种高灵敏的电化学生物传感器(图5C)。Pb2+的存在激活了DNA酶的活性,故底物链被切割为两段并游离出来。游离出来的标记有生物素的底物链片段可与组装在金电极表面的捕获探针杂交,然后标记有链霉亲和素的PbS量子点通过亲和素与生物素的特异性识别作用被固定在电极表面,最后通过电化学溶出法就可以测定Pb2+含量,其检出限为0.6 nmol/L。Tang等[61]将量子点作为标记物并利用滚环扩增原理设计了一种可高灵敏检测Pb2+的电化学型DNA酶传感器。此外,量子点还可以与脱氧核酶结合作为一个信号放大基团用于其它物质的检测。如Zhang等[62]将PbS量子点标记在二抗上,有目标物AFP存在时,PbS量子点就可以通过夹心结构固定在标记有一抗的微孔板中。然后量子点中的Pb2+可以通过酸溶出法释放出来,被释放的Pb2+就可以激活组装在电极表面的DNA酶的活性并使底物链被切割,导致标记在酶链上的电化学活性物质二茂铁靠近电极并产生强的电化学信号,从而实现了AFP的高灵敏检测。
5 脱氧核酶-磁性纳米颗粒复合物在生物传感中的应用
磁性纳米材料是20世纪80年代出现的一种新型纳米材料,由于其具有优异的磁学性能、良好的生物相容性以及简单的表面修饰等优点,被广泛应用于临床诊断、磁共振成像、细胞分离以及生物检测等各个领域[63~65]。磁性纳米颗粒与脱氧核酶结合在生物传感中也得到了广泛应用[66~71]。Nie等[66]基于流式细胞计数法构建了一种DNA酶生物传感器, 用于Pb2+的检测(图6A)。其中GR-5DNAzyme的一端标记有荧光团,另一端则共价修饰于磁珠上,加入Pb2+后,两端标记有猝灭团的底物链被切割并与酶链分离,致使传感器的荧光恢复,由此可以定量检测Pb2+。
使用流式细胞计数法可以降低Pb2+检测过程中光散射造成的干扰。Ge等[67]将捕获探针共价修饰于磁珠上,Cu2+的存在使底物链被切割为两段,其中的一段底物片段就可以作为催化剂引发两条信号探针在磁珠上的自组装,
图6 基于脱氧核酶与磁性纳米颗粒结合的生物传感器检测原理图[66,68]
Fig.6 Schematic illustration of DNAzyme-magnetic bead based biosensors[66,68]
最后通过SYBR Green Ⅰ嵌入核酸双链实现Cu2+的无酶、无标记荧光放大检测。该传感器设计简单、花费低廉且灵敏度高,其检出限是12.8 pmol/L。除了利用RNA切割型脱氧核酶设计生物传感器外,也可基于G四聚体脱氧核酶的信号放大作用设计一些高灵敏度的传感器。Du等[68]在磁性纳米颗粒上共价修饰了可卡因核酸适配体的一个片段,当有可卡因存在时,与另外一条连接有G四聚体脱氧核酶的核酸适配体片段形成夹心复合物,然后通过磁性分离与脱氧核酶催化TMB显色来比色检测可卡因(图6B)。Tang等[69]则依据上述夹心法原理并采用滚环放大策略扩增G四聚体脱氧核酶片段最后实现PDGF的超灵敏检测。磁性纳米颗粒的磁性富集和分离功能可以有效的降低背景信号,实现目标物的高灵敏检测,此外还可以减少实际样品中非目标物的干扰。
6 脱氧核酶-其它纳米材料复合物在生物传感中的应用
脱氧核酶除了与上述几种纳米材料结合外,其它纳米材料如水凝胶、树枝状DNA纳米材料等也可以用来
图7 基于脱氧核酶与水凝胶结合的生物传感器检测原理图 [73]
Fig.7 Schematic illustration of DNAzyme-hydrogel-based biosensors [73]
与DNA酶结合发展各种生物传感器[72~74]。例如Lin等[72]利用DNA酶为水凝胶的交联剂构建了可检测金属离子的传感平台(图7)。当存在Cu2+时,水凝胶从凝胶状态变为溶胶状态,此时包裹在凝胶中的纳米金被释放出来,因此可以根据纳米金的释放量来比色检测Cu2+。本研究组[73]将DNA酶通过碱基互补组装在DNA树枝状纳米材料中,并构建了生物相容性好、膜穿透能力强、高灵敏、高选择性的荧光纳米探针,实现了细胞内组氨酸的成像检测。
7 总结与展望
生命科学与纳米材料是21世纪最前沿的两大学科,纳米材料的介入为生物传感器的发展提供了无穷的空间。目前基于脱氧核酶和纳米材料结合的生物传感器已经得到快速的发展,并深入到多个分析领域,展现出独特的优势。但是要进一步将其用于临床和实际检测,还有许多方面有待优化。
首先,当前大部分文献报道的传感器都仅限于在缓冲溶液中检测,没有真正应用到实际样品及生物体内。事实上,在实际环境检测和医疗诊断中其样品成分非常复杂,故DNA酶传感器在实际应用中其性能容易受生物介质的干扰。因此,可以考虑将DNA酶组装在介孔纳米材料的内部,从而进一步减少生物样品分析中核酸酶等非目标组分的干扰。其次,将DNA酶传感器用于体内生物分子检测时,纳米材料的生物安全性是需要考虑的问题。因此需要研发新型安全无毒的纳米材料作为DNA酶的运载体。DNA作为一种天然生物分子,具有良好的生物相容性,将DNA纳米材料如四面体DNA和DNA纳米花等作纳米载体用于构建DNA酶传感器可以降低载体对细胞或组织的毒性,从而推进传感器真正应用于临床检测。最后,目前用于传感器设计的DNA酶主要是RNA切割型脱氧核酶与G四聚体脱氧核酶,其检测目标范围有限。如果研究和筛选针对更多特定靶分子的脱氧核酶并将其用于传感器的构建,可以大大拓展脱氧核酶传感器的适用范围。随着研究的不断深入,脱氧核酶与纳米材料结合的生物传感器将被广泛应用于临床诊断、环境检测与食品安全等与生活息息相关的领域。
References
1 Eychmuller A. J. Phys. Chem. B, 2000, 104(28): 6514-6528
2 Ball P,Garwin L. Nature, 1992, 355(6363): 761-766
3 Cavucchi P E, Silsbee R H. Phys. Rev. Lett., 1984, 52(16): 1453-1456
4 Penn S G, He L, Natan M J. Curr. Opin. Chem. Biol., 2003, 7(5): 609-615
5 ZHANG Yu-Wei, XU Wei-Lin. Chinese J. Anal. Chem., 2013, 41(12): 1932-1938
张玉微, 徐维林. 分析化学, 2013, 41(12): 1932-1938
6 Singh A K, Flouders A W, Volponi J V, Ashley C S, Wally K, Schoeniger J S. Biosen. Bioelectron., 1999, 14(8-9): 703-713
7 Bauer G, Pittner F, Schalkhammer T. Microchim. Acta, 1999, 131(1-2): 107-114
8 Cuenoud B, Szostak J W. Nature, 1995, 375(15): 611-614
9 Santoro S W, Joyce G F. Proc. Natl. Acad. Sci. USA, 1997, 94(9): 4262-4266
10 ZHAO Yong-Xi, QI Lin, YANG Wei-Jun, WEI Shuai, WANG Ya-Ling. Chinese J. Anal.Chem., 2012, 40(8): 1236-1240
赵永席, 齐 林, 杨卫军, 魏 帅, 王亚玲. 分析化学, 2012, 40(8): 1236-1240
11 Breaker R R. Nat. Biotechnol., 1997, 15(5): 427-431
12 Breaker R R, Joyce G F. Chem. Biol., 1994, 1(4): 223-229
13 Brown A K, Li J,Pavot C M B. Lu Y. Biochemistry, 2003, 42(23): 7152-7161
14 Santoro S W, Joyce G F,Sakthivel K. J. Am. Chem. Soc., 2000, 122(11): 2433-2439
15 Carmi N, Balkhi H R, Breaker R R. Proc. Natl. Acad. Sci. USA, 1998, 95(5): 2233-2237
16 Faulhammer D, Famulok M. Angew. Chem. Int. Ed., 1996, 35(5): 2837-2841
17 Gao L, Li L L, Wang X L, Wu P W, Cao Y, Liang B, Li X, Lin Y W, Lu Y, Guo X F. Chem. Sci., 2015, 6: 2469-2473
18 Saadaoui M, Fernandez I, Sanchez A, Diez P, Campuzano S, Raouafi N, Pingarron J, Villalonga R. Electrochem. Commun. 2015, 58: 57-61
19 Yim T J, Liu J W, Lu Y, Kane R S, Dordick J S. J. Am. Chem. Soc., 2005, 127(35): 12200-12201
20 Warashina M, Kuwabara T, Nakamatsu Y, Taira K. Chem. Biol, 1999, 6(4): 237-250
21 Fan , Zhao Z L, Yan G B, Zhang X B, Yang C, Meng H M, Chen Z, Liu H, Tan W H. Angew. Chem.Int. Edit, 2015, 127(16): 4883-4887
22 Li J W, Lu Y. J. Am. Chem. Soc., 2000, 122(42): 10466-10467
23 Liu J W, Lu Y. Anal. Chem., 2003, 75(23): 6666-6672
24 Chiuman W, Li Y. Nuleic. Acids Res., 2007, 35(2): 401-405
25 Daniel M C, Astruc D. Chem. Rev., 2003, 104(1): 293-346
26 Zhang C, Zhang Z, Yu B, Shi J, Zhang X. Anal. Chem., 2002, 74(1): 96-99
27 Raj C R, Okajima T, Ohsaka T. J. Electroanal. Chem., 2003, 543(2): 127-133
28 Liu S Q, Ju H X. Biosens. Bioelectron., 2003, 19(3): 177-183
29 Liu J W, Lu Y. J. Am. Chem. Soc., 2003, 125(22): 6642-6643
30 Liu J W, Lu Y. J. Am. Chem. Soc., 2004, 126(39): 12298-12305
31 Liu J W, Lu Y. J. Am. Chem. Soc., 2005, 127(36): 12677-12683
32 Lee J H, Wang Z D, Liu J W, Lu Y. J. Am. Chem. Soc., 2008, 130(43): 14217-14226
33 Wei H, Li B L, Li J, Dong S J, Wang E K. Nanotechnology, 2008, 19(9): 095501
34 Liu J W, Lu Y. Chem. Commun., 2007, 46: 4872-4874
35 Wang H B, Wang L, Huang K J, Xu S P, Wang H Q. Wang L L, Liu Y M. New J. Chem., 2013, 37(8): 2557-2563
36 Malashikhina N, Pavlov V. Biosens Bioelectron., 2012, 33(1): 241-246
37 Yin B C, Zuo P, Huo H, Zhong X H, Ye B C. Anal. Biochem., 2010, 401(1): 47-52
38 Wu P, Hwang K, Lan T, Lu Y. J. Am. Chem. Soc., 2013, 135(14), 5254-5257
39 Sun Y H, Kong R M, Lu D Q, Zhang X B, Tan W H, Shen G L, Yu R Q. Chem. Commun., 2011, 47: 3840-3842
40 Shen L, Chen Z, Li Y H, He S L, Xie S B, Xu X D, Liang Z W, Meng X, Li Q, Zhu Z W, Li M X, Le C, Shao Y H. Anal. Chem., 2008, 80(16): 6323-6328
41 Li L D, Chen Z B, Zhao H T, Guo L. Biosens. Bioelectron., 2011, 26(5): 2781-2785
42 Pelossof G, Vered R T, Willner I. Anal. Chem., 2012, 84(8), 3703-3709
43 Geim A K, Novoselov K S. Nat. Mater., 2007, 6(3): 183-191
44 Wen Y Q, Peng C, Li D, Zhuo L, He S J, Wang L H, Huang Q, Xu Q H, Fan C H. Chem. Commun., 2011, 47: 6278-6280
45 Zhao X H, Kong R M, Zhang X B, Meng H M, Liu W N, Tan W H, Shen G L, Yu R Q. Anal. Chem., 2011, 83(13): 5062-5066
46 Yu Y, Liu Y, Zhen S J, Huang C Z. Chem. Commun., 2013, 49: 1942-1944
47 Liu M, Zhao H M, Chen S, Yu H T, Zhang Y B, Quan X. Chem. Commun., 2011, 47: 7749-7751
48 Kim S C, Ryoo S R, Na H K, Kim Y K, Choi B S, Lee Y H, Kim D E, Min D H. Chem. Commun., 2013, 49: 8241-8243
59 Liu M, Zhao H M, Chen S, Yu H T, Zhang Y B, Quan X. Biosens Bioelectron., 2011, 26(10): 4111-4116
50 Luo M, Chen X, Zhou G H, Xiang X, Chen L, Ji X H, He Z K. Chem. Commun., 2012, 48: 1126-1128
51 Yuan Y L, Liu G P, Yuan R, Chai Y Q, Gan X X, Bai L J. Biosens. Bioelectron., 2013, 42(15): 474-480
52 Xie S B, Chai Y Q, Yuan R, Bai L J, Yuan Y L, Wang Y. Anal. Chim. Acta, 2012, 755(28): 46-53
53 Smith A M, Gao X H, Nie S M. Photochem. Photobiol., 2004, 80(3): 377-385
54 Bailey R E, Nie S M. J. Am. Chem. Soc., 2003, 125(23): 7100-7106
55 Hines M A, Scholes G D. Adv. Mater., 2003, 15(21): 1842-1849
56 Jaiswal J K, Simon S M. Trends Cell Biol., 2004, 14(9): 497-504
57 Wu C S, Khaing O M K, Fan X D. ACS Nano., 2010, 4(10): 5897-5904
58 Sharon E, Freeman R,Willner I. Anal. Chem., 2010, 82(17): 7073-7077
69 Hu R, Liu T, Zhang X B, Yang Y H, Chen T, Wu C C, Liu Y, Zhu G Z, Huan S Y, Fu T, Tan W H. Anal. Chem., 2015, 87 (15): 7746-7753
60 Zhang H X, Jiang B Y, Xiang Y, Su J, Chai Y Q, Yuan R. Biosens. Bioelectron., 2011, 28 (1): 135-138
61 Tang S R, Tong P, Li H, Tang J, Zhang L. Biosens. Bioelectron., 2013, 42: 608-611
62 Zhang B, Liu B Q, Zhou J, Tang J, Tang D P. ACS Appl. Mater. Interfaces, 2013, 5(10): 4479-4485
63 Pankhurst Q A, Connolly J, Jones S K. Dobson J. J. Phys. D: Appl. Phys., 2003, 36: 167-181
64 Gupta A K, Gupta M. Biomaterials, 2005, 26(18): 3995-4021
65 Harisinghani M G, Barentsz J, Hahn P F. Deserno W M, Tabatabaei S, Kaa C H, Rosette J, Weissleder R. N Engl. J. Med., 2003, 348: 2491-2499
66 Nie D D, Wu H Y, Zheng Q S, Guo L Q, Ye P R, Hao Y N, Fu F F, Guo Y H. Chem. Commun., 2012, 48: 1150-1152
67 Ge C C, Chen J H, Wu W, Fang Z Y, Chen L B, Liu Q, Wang L, Xing X R, Zeng L W. Analyst, 2013, 138(17): 4737-4740
68 Du Y, Li B L, Guo S J, Zhou Z X, Zhou M, Wang E K, Dong S J. Analyst, 2011, 136(3): 493-497
69 Tang L H, Liu Y, Ali M M, Kang D K, Zhao W A, Li J H. Anal. Chem., 2012, 84(11): 4711-4717
70 Zhuang J Y, Fu L B, Xu M D, Zhou Q, Chen G N, Tang D P. Biosens. Bioelectron., 2013, 45: 52-57
71 Bi S, Li L, Zhang S S. Anal. Chem., 2010, 82(22): 9447-9454
72 Lin H X, Zou Y, Huang Y S, Chen J, Zhang W Y, Zhuang Z X, Gareth J, Yang C Y. Chem. Commun., 2011, 47: 9312-9314
73 Meng H M, Zhang X B, Lv Y F, Zhao Z L, Wang N N, Fu T, Fan H H, Liang H, Qiu L P, Zhu G Z, Tan W H. ACS Nano, 2014, 8(6): 6171-6181
74 Chien M P, Thomposon M P, Gianneschi N C. Chem. Commun., 2011, 47: 167-169
- 传统媒体如何打造时尚活动
- 停刊事件中报业的危机传播策略
- 2001-2019年央视春晚小品的叙事学分析
- 《南方》杂志社开发战“疫”机器人
- 融合生态下如何发挥媒介财务管理的助力作用
- 跨文化视野下《嘎达梅林》的割裂化表达
- 美国电影中新闻专业主义的构建研究
- 媒体融合背景下“全民阅读”建设理路探析
- “两微一端”视野下的电视转型
- 媒体应向滥食野生动物陋习说“不”
- 县级融媒体中心建设途径分析
- 主流纸媒如何彰显文化担当
- 性犯罪新闻报道的价值偏移与伦理规范
- 论社交媒体公众号的权利、责任及言论规范
- 社交媒体与公众健康运动文献综述
- 新闻学概论课程思政育人的探索与实践研究
- 新媒体环境下少数民族地区新闻人才的能力结构变迁研究
- “铁脚板”下出新闻
- 2019中国媒体十大流行语揭榜
- 600米矿井下的洗礼
- 发现让人惊叹的新闻角度
- 全媒体时代党报记者如何做好“社会活动家”
- 广播新闻如何讲好中国故事
- 用好“短视频”做优融合类报道
- VR技术对新闻产品创新的影响
- spermicides
- sperms
- sperm whale
- sperm whales
- spermy
- spew
- spewed
- spewer
- spewers
- spewing
- spewings
- spews
- speˌcific risk
- sphere
- sphered
- sphereless
- spherelike
- spheres
- spherical
- sphericalities
- spherically
- sphericities
- spherify
- sphering
- spheroid
- 拘拘缩缩
- 拘括
- 拘拿
- 拘挛
- 拘挛之见
- 拘挛儿
- 拘挛守常
- 拘挛补纳
- 拘捕
- 拘捕人的公文
- 拘捕囚禁
- 拘捕审问
- 拘捕弹劾
- 拘捕押送
- 拘捕拷打
- 拘捕管教
- 拘捕问罪
- 拘控
- 拘提
- 拘摄
- 拘摄抽收
- 拘收
- 拘教
- 拘文
- 拘文执法