米雪?都修波?常琳琳?谭泽圆?马丙祥?郑宏
【摘要】糖原累积症IXa型(GSD IXa) 是由PHKA2基因杂合突变引起的X连锁隐性遗传病。该文报道1例GSD IXa型患儿的临床资料并复习相关文献。该例患儿男,9月龄,因“运动发育落后、发现肝酶升高3个月”就诊,临床表现为翻身不灵活,不能独坐,ALT 84.0 U/L,AST 120.2 U/L,AST同工酶33.6 U/L,总蛋白57.9 g/L,白蛋白42.1 g/L,球蛋白15.8 g/L,彩色多普勒超声检查(彩超)示肝脏增大。基因检测显示患儿PHKA2基因c.3143C>T(p.T1048M)半合子突变,突变来自于杂合子母亲,为错义突变。结合患儿临床特征和基因突变测序结果,确诊为GSD IXa型。遂予患儿口服生玉米淀粉每日4次、餐间服用,予口服葡醛内酯片、复方甘草酸苷片等护肝。5个月后随访,患儿肝功能较前明显改善,复查ALT 40.0 U/L,未复查肝脏彩超,运动发育基本与同龄正常儿童相同。
【关键词】糖原累积症IXa型;PHKA2基因;发育落后;肝脏肿大;肝酶高
PHKA2-related glycogen storage disease type IXa: a case report Mi Xue, Du Xiubo, Chang Linlin, Tan Zeyuan, Ma Bingxiang, Zheng Hong. Henan University of Chinese Medicine, Zhengzhou 450046, China
Corresponding author, Du Xiubo, E-mail: 3132939789@ qq. com
【Abstract】Glycogen storage syndrome type IXa (GSD IXa) is an X-linked recessive genetic disease caused by heterozygous mutation of PHKA2 gene. In this article, clinical data of a child with GSD IXa were reported and related literature review was conducted. The boy aged 9 months was admitted to our hospital due to delayed development and hypertransaminasemia for 3 months. As for clinical manifestations, the boy could not turn over agilely or sit stably by himself. The ALT level was 84.0 U/L, 120.2 U/L for AST, 33.6 U/L
for AST isoenzyme, 57.9 g/L for total protein, 42.1 g/L for albumin and 15.8 g/L for globulin, respectively. Color Doppler ultrasound showed liver enlargement. Genetic testing showed that the child had a hemizygous mutation of c.3143C > T (p.T1048M) in the PHKA2 gene, it was a missense mutation from a heterozygous mother. The diagnosis of GSD IXa was confirmed combined with the clinical characteristics and gene mutation sequencing results. The child was orally given with raw corn starch four times a day between meals, and glucurolactone tablets and compound glycyrrhizin tablets to protect the liver. After 5-month follow-up, the liver function of the child was significantly improved. The ALT level was detected as 40.0 U/L. Color Doppler ultrasound was not performed. Physical development of the child was basically the same as normal counterparts of the same age.
【Key words】Glycogen storage disease type IXa;PHKA2 gene;Hypoevolutism;Hepatomegaly;
Hypertransaminasemia
糖原累積症(GSD)是一种糖原代谢异常的遗传性疾病。由于本病仅靠临床表现不易确诊,且多数医师对本病认识不足,故易误诊漏诊。笔者收治1例GSD患儿,该患儿运动发育落后,肝功能损伤,护肝治疗效果不佳,辗转几所医院未明确病因,经基因检测后确诊为GSD IXa型,现将该例报道如下。
病例资料
一、主诉及病史
患儿男,9月龄,因“运动发育落后,发现肝酶升高3个月”于2018年8月28日入河南中医药大学第一附属医院。患儿6月龄时因翻身不灵活、不能独坐于外院住院治疗,行肝功能检查示ALT 75.0 U/L,AST 105.0 U/L,总蛋白57.5 g/L,白蛋白35.6 g/L;血氨基酸及肉碱谱分析:丙酰基肉碱降低,乙基丙二酸增高;尿有机酸分析示己二酸及辛二酸增高,可能与脂肪酸氧化有关,3-羟基丁酸增高,提示酮尿;行表面肌电图检查示肱二头肌、内收肌信号活动稍弱;脑干听觉诱发电位示左侧脑干未见异常,右侧70 dB各波潜伏期及峰间值均正常,各波波幅与对侧相比均略低,双侧阈值30 dB;视觉诱发电位未见异常;头颅MRI示髓鞘化落后于同龄儿。外院诊断为运动发育落后原因待查,予口服左卡尼汀、葡醛内酯片、维生素C片及综合康复治疗,半个月后复查肝功能示ALT 93.0 U/L,AST 137.0 U/L,为进一步治疗转至河南中医药大学第一附属医院就诊。患儿是头胎,足月经剖宫产产出,出生体质量4550 g,出生时无缺氧窒息等,无生理性黄疸,母亲孕晚期出现1次尿葡萄糖(+),经饮食控制后恢复正常,孕后期胎动减少,吸氧后胎动恢复正常,余无异常。患儿父母均身体健康,非近亲婚配,否认有家族遗传病史。
二、体格检查
体质量7.3 kg,身长70 cm,发育落后,营养不良貌,皮下脂肪菲薄,皮肤弹性差,褶皱多,腰背及臀部可见大片形状不规则蒙古斑,肋缘外翻,呈“O”型腿。肝脏可触及肿大,质韧,边缘圆钝,肝浊音界增大,无肝区叩击痛。专科查体:头发稀疏、纤细,头围44 cm,前囟平软,面积约3 cm×3 cm,眼距2.7 cm,小下颌,长人中,追视追听正常,可逗笑,能笑出声;肩肘关节稍有抵抗,股角120°,腘角150°,足背屈角60°,踝关节抵抗;竖头稳,翻身灵活;弓背坐;俯卧位肘支撑,抬头90°;仰卧位肢体对称,有主动抓物意识,可对指捏取;立位双下肢可支持身体,无主动迈步意识;Vojta反射,拉起头后垂,能主动用力;原始反射,摩罗(Moro)反射呈伸展相,非对称性紧张性颈反射(-),对称性紧张性颈反射(-),迷路紧张反射(-),侧弯反射(-),手、足把握(-),落伞反射可引出;膝腱反射(++++),踝阵挛(-),巴宾斯基征(+)。
三、实验室及辅助检查
尿、粪常规未见异常。血常规示血红蛋白106 g/L,红细胞3.98×1012/L,红细胞平均体积80.1 fl,平均血红蛋白量26.5 pg,血小板670×109/L白细胞8.7×109/L。ALT 84.0 U/L,AST 120.2 U/L,AST同工酶33.6 U/L,总蛋白57.9 g/L,白蛋白42.1 g/L,总胆红素6.0 μmol/L、直接胆红素1.7 μmol/L、间接胆红素4.3 μmol/L,球蛋白15.8 g/L,肌酸激酶62.0 U/L,CK-MB 20.0 U/L;血糖3.79 mmol/L(参考值3.90 ~ 6.10 mmol/L),甘油三酯0.56 mmol/L(参考值0 ~ 1.70 mmol/L);血同型半胱氨酸未见异常。患儿对食物不耐受,鸡蛋高度敏感,牛奶、大米、小麦轻度敏感;连续1周监测空腹及餐前血糖为3.8 ~ 6.4 mmol/L。颈胸腰椎X线片未见异常。彩色多普勒超声检查(彩超)示心脏未见异常;副脾;肝右叶斜径73 mm,肋下30 mm,大于正常肝脏;泌尿系统未见异常。心电图示窦性心律不齐。
四、基因检测情况
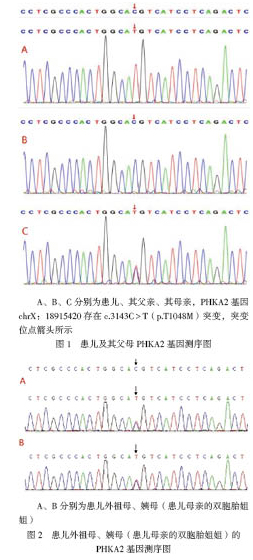
结合患儿临床症状及实验室检查结果,疑为GSD,家属签署知情同意书,取患儿及其父母静脉血送检,采用全外显子及Sanger測序验证进行相关基因分析。结果显示患儿PHKA2基因存在1个突变点c.3143(exon 30)C > T(p.T1048M),位于第30外显子,为致病性突变。查阅单核苷酸多态性数据库(dbSNP)、千人基因组、千人南方、千人北方、外显子集成联合数据库(ExAC)、ExAC东亚等均未见收录,根据美国医学遗传学与基因组学学会(ACMG)变异解读指南进行生物学致病等级分析,位点c.3143C > T(p.T1048M)错义变异位于深入研究的无良性变异的外显子功能域,属于低频变异,预测无证据表明变异对基因(基因产物)有影响,保守性及蛋白结构预测无害;该变异先证者为半合子,符合X染色体隐性遗传疾病发病机制,先证者及其家庭成员表型及其基因型符合共分离,变异支持致病。综上所述,提示本例患儿发病与PHKA2基因变异的危害性与表型存在相关性,最终结合临床确诊为GSD IXa型。患儿家族基因测序结果见图1、2。
五、治疗及随访
给予患儿口服生玉米淀粉(患儿体质量7.3 kg,故给予生玉米淀粉15 g/d),每日4次,餐间服用;予口服葡醛内酯片、复方甘草酸苷片等护肝。出院5个月后随访,患儿肝功能较前明显改善,ALT 40.0 U/L,未复查肝脏彩超,运动发育情况基本与同龄正常儿童相同。
讨论
GSD又被称为肝糖原累积病,是由糖原代谢相关酶缺乏或者活性降低引起的一组临床和遗传异质性疾病,可导致糖原在多个组织脏器中累积,以肝脏为主要受累器官,根据酶、受累组织不同和发现时间的先后,GSD被分为13种类型[1]。GSD IX是糖原累积症中最常见的类型,约占所有GSD的25%,发病率约为1/100 000[2]。已知GSD IX致病基因主要有3种,分别为PHKA2、PHKB、PHKG2,由此可将GSD分为3种类型,分别为GSD IXa、GSD IXb和GSD IXc。GSD IXa是GSD IX中最常见的类型,由PHKA2突变引起,磷酸化酶激酶(PHK)由α、β、γ和δ 4个亚基组成,不同基因编码不同的亚基,PHKA2基因位于X染色体的长臂上(Xp22.2-22.1),编码PHK的α亚基,包括33个外显子,PHKA2基因突变是导致PHK缺乏的常见原因,约占75%[3-4]。PHK缺乏引起的GSD主要临床症状为肝脏增大、肝酶升高、低血糖、高血脂、酮症、生长发育迟缓和矮小[5]。少数患者可有肝纤维化改变、特应性皮炎、反复性腹泻、肝硬化和罕见的迟发性神经系统症状,也有患者以间断便血为首发症状[6-10]。检索人类基因突变数据库截至2019年10月报道的GSD IXa病例有134例,症状以肝酶升高、肝脏增大、低血糖多见,收录的PHKA2基因突变包括错义、缺失、插入、无义、剪接突变,这些症状与PHKA2基因突变所致表型相符,与GSD其他类型相比,GSD IXa的症状较轻,可随着年龄的增长而改善,一般预后良好,但其临床症状表现严重程度不一,即使无低血糖和酮症表现,也要常规监测葡萄糖和酮水平,因为未检测到的低血糖和酮症会影响患者生长发育,需及时干预[11-12]。
GSD IXa的治疗主要采用生玉米淀粉,或者采用高蛋白食物维持血糖、改善肝功能及对症治疗[13-15]。但这种治疗对疾病并发症的预防作用尚不明确,应定期检测患儿肝脏及心脏各项指标,防止发生并发症[16]。目前在我国已被报道的以运动发育迟缓为主要表现而确诊的患儿有2例[17]。本例患儿亦以发育迟缓为主诉就诊,经过基因测序发现c.3143C > T(p.T1048M)突变,新的突变位于PHKA2第30号外显子上,先证者的突变来自其母亲。验证先证者外祖母及姨母基因,结果均为携带者,患儿母亲、外祖母及姨母并未受到影响,符合该病X连锁隐性遗传的方式,但也有女性发病的文献报道[18]。患儿初始表现为运动发育落后于同龄儿,进一步检查后发现肝酶异常及肝脏增大,有可能是肝酶升高出现较早但未行检查所致,连续监测空腹血糖未见异常,考虑与患儿进食母乳频繁、空腹时间较短,糖异生途径受影响小有关,明确诊断后即采用护肝药物及生玉米淀粉以改善肝功能、预防低血糖,同时定期检测肝脏彩超。
GSD具有临床和遗传异质性,即使同一基因突变,临床症状也不尽相同,不同基因突变也可能出现同样症状,加之GSD IXa症状较轻,不典型,所以诊断该病不易,有些患儿在出现肝脏肿大、低血糖之前,先出现原因不明的生长发育迟缓和反复腹泻,也会给本病的诊断增加难度,若不及时明确诊断、治疗,会影响患儿正常发育。传统的依赖肝脏、肌肉活组织检查或血液生化结果的方式有创且并发症多,随着基因检测技术的发展,疑为GSD时应尽早进行基因检测,以助尽早明确诊断、进行针对性治疗。
参 考 文 献
[1] 顾学范, 江载芳, 申昆玲, 沈颖.褚福堂实用儿科学.8版.北京:人民卫生出版社, 2015:2263.
[2] Maichele AJ, Burwinkel B, Maire I, S?vik O, Kilimann MW. Mutations in the testis/liver isoform of the phosphorylase kinase gamma subunit (PHKG2) cause autosomal liver glycogenosis in the gsd rat and in humans. Nat Genet,1996,14(3):337-340.
[3] Johnson AO, Goldstein JL, Bali D. Glycogen storage disease type IX: novel PHKA2 missense mutation and cirrhosis. J Pediatr Gastroenterol Nutr,2012,55(1):90-92.
[4] Bali DS, Goldstein JL, Fredrickson K, Austin S, Pendyal S, Rehder C, Kishnani PS. Clinical and molecular variability in patients with PHKA2 variants and liver phosphorylase b kinase deficiency. JIMD Rep, 2017,37:63-72.
[5] Zhu Q, Wen XY, Zhang MY, Jin QL, Niu JQ. Mutation in PHKA2 leading to childhood glycogen storage disease type IXa: a case report and literature review. Medicine (Baltimore),2019,98(46):e17775.
[6] Kim TH, Kim KY, Kim MJ, Seong MW, Park SS, Moon JS, Ko JS. Molecular diagnosis of glycogen storage disease type IX using a glycogen storage disease gene panel. Eur J Med Genet,2020,63(6):103921.
[7] Achouitar S, Goldstein JL, Mohamed M, Austin S, Boyette K, Blanpain FM, Rehder CW, Kishnani PS, Wortmann SB, den Heijer M, Lefeber DJ, Wevers RA, Bali DS, Morava E. Common mutation in the PHKA2 gene with variable phenotype in patients with liver phosphorylase b kinase deficiency. Mol Genet Metab,2011,104(4):691-694.
[8] Tsilianidis LA, Fiske LM, Siegel S, Lumpkin C, Hoyt K, Wasserstein M, Weinstein DA. Aggressive therapy improves cirrhosis in glycogen storage disease type IX. Mol Genet Metab,2013,109(2):179-182.
[9] Smith C,Care4Rare Canada Consortium, Dicaire MJ, Brais B, La Piana R. Neurological involvement in glycogen storage disease type IXa due to PHKA2 mutation. Can J Neurol Sci,2020,47(3):400-403.
[10] 孙曼青,陆文丽,王伟,董治亚,肖园,倪继红,王德芬.以间断便血为首发表现的PHKA2基因突变致糖原累积病Ⅸa 1例报告.中国实用儿科杂志,2020,35(3):246-248.
[11] Zhang J, Yuan Y, Ma M, Liu Y, Zhang W, Yao F, Qiu Z. Clinical and genetic characteristics of 17 Chinese patients with glycogen storage disease type IXa. Gene,2017,627:149-156.
[12] Kim JA, Kim JH, Lee BH, Kim GH, Shin YS, Yoo HW, Kim KM. Clinical, Biochemical, and genetic characterization of glycogen storage type IX in a child with asymptomatic hepatomegaly. Pediatr Gastroenterol Hepatol Nutr,2015,18(2):138-143.
[13] 李春微,于康,李融融,李明,張敏杰.糖原累积症的医学营养治疗:病例报告与文献综述.营养学报,2014,36(6):594-602,630.
[14] 李静.糖原累积症患儿临床特征与治疗.临床检验杂志(电子版),2018,7(3):440.
[15] 王璞,董漪,徐志强,陈大为,王福川,甘雨,王丽旻,闫建国,曹丽丽,李爱芹,朱世殊,张敏.糖原累积症Ⅸ型12例临床、病理特点及基因突变位点分析.肝脏,2018,23(9):764-768.
[16] Roscher A, Patel J, Hewson S, Nagy L, Feigenbaum A, Kronick J, Raiman J, Schulze A, Siriwardena K, Mercimek-Mahmutoglu S. The natural history of glycogen storage disease types VI and IX: Long-term outcome from the largest metabolic center in Canada. Mol Genet Metab,2014,113(3):171-176.
[17] Fu J, Wang T, Xiao X. A novel PHKA2 mutation in a Chinese child with glycogen storage disease type IXa: a case report and literature review. BMC Med Genet,2019,20(1):56.
[18] Cho SY, Lam CW, Tong SF, Siu WK. X-linked glycogen storage disease IXa manifested in a female carrier due to skewed X chromosome inactivation. Clin Chim Acta,2013,426:75-78.
(收稿日期:2020-07-31)
(本文编辑:洪悦民)
- 高中生二语语义趋向知识习得研究
- 课程思政体系下的大学英语教学探索
- 基于超星学习通平台实现大学英语机考的实践探究
- 高职英语专业学生思辨能力培养的行动研究
- 高职院校听障生英语课堂有效教学对策探析
- 混合式教学模式在高职英语教学中的应用策略
- 浅谈如何提高学生英语听力水平
- 基于翻转课堂的高职英语口语教学实证研究
- 基于全人教育理念下的大学英语教学探究
- 高职英语课堂的思政教育渗透研究
- 中职英语教师提高职业素养之我见
- 四位一体监控体系在高职英语网络自主学习的应用探讨
- 校企合作背景下的高职高专英语人才培养模式探究
- 基于翻转课堂的高职英语新型混合教学策略分析
- 情境教学法在中职英语视听说教学中的应用研究
- 技工院校英语教师实践教学探索与研究
- 浅谈网络“微环境”下英美文学教学
- 论“电子档案袋”对高职英语教学的促进作用
- 读英文原著对大学生英语学习的帮助
- 高职英语教育与职业技能教育的有效结合研究
- 世界技能大赛英语培训必要性的研究
- 从高职英语教学现状谈高职英语有效教学策略分析
- 借助网络力量,优化英语作业
- 中职幼师英语口语策略浅析
- 如何提高学生英语阅读能力
- inequity
- inequity's
- iner
- inert
- inertia
- inertiae
- inertias
- inertion
- inertly
- inertness
- inertnesses
- inerts
- inescapability
- inescapable
- inescapableness
- inescapablenesses
- inescapably
- inestimabilities
- inestimability ,inestimableness
- inestimable
- inestimablenesses
- inestimably
- in-event-of
- in every sense
- in every way/respect/detail
- 无两
- 无丧不掉泪,无仇难开刀
- 无中挑有
- 无中生有
- 无中生有地侮辱
- 无中生有地引诱他人入罪
- 无中生有地挑拨
- 无中生有地罗织罪名
- 无中生有地说他人坏话
- 无中生有地说好
- 无中生有的事
- 无中生有的指控
- 无中生有的毁谤
- 无中生有,凭空胡诌
- 无中生有,荒诞离奇
- 无中生有,骗人说谎
- 无中觅有
- 无为
- 无为牛后,宁作鸡前
- 无为而成
- 无为而极安宁的境界
- 无为而治
- 无为而治、天下太平的局面
- 无为而治,社会安定,政事很少
- 无为集