表面增强拉曼光谱结合乘子效应模型对血浆和药片中甲巯咪唑的定量检测
胡敏 陈增萍 陈瑶 石彩霞 俞汝勤
摘 要 将表面增强拉曼散射(Surface-enhanced raman scattering,SERS)技术与乘子效应模型结合,采用2-巯基异烟酸作为内标,以银纳米颗粒为SERS增强基底,对血浆和药片样本中甲巯咪唑的进行准确的定量分析。实验结果表明,本方法对血浆样本中甲巯咪唑的平均相对预测误差为5.1%,检出限为32 nmol/L;对药片中甲巯咪唑的定量结果与LC-MS/MS方法基本一致,其加标回收率在93.3%~110.9%之间。
关键词 表面增强拉曼光谱; 甲巯咪唑; 2-巯基异烟酸; 乘子效应模型; 定量分析
1 引 言
甲巯咪唑为抗甲状腺药物,一般用于各种类型的甲状腺功能亢进症【1,2】的治疗。但该药物的使用不当或者过量使用会产生一定的副作用,轻则出现粒细胞减少,脱发、皮炎等,重则可能导致中毒性肝损伤。因此,必须在甲巯咪唑的生产和使用过程进行严格的定量监控。目前,甲巯咪唑的定量分析方法有液相色谱法【3,4】、气相色谱-质谱法【5,6】、电化学方法【7,8】和间接流动注射法【9】等。但是,这些方法均具有一定的局限性:需要对复杂实际样品进行繁琐费时的预处理,或者所需仪器价格昂贵且体积庞大,不适用于大量样品的快速在线分析。因此, 建立一种新型简便、快速、灵敏的甲巯咪唑的定量方法仍具有较重要的现实意义。
表面增强拉曼散射技术(Surface-enhanced raman scattering,SERS)具有光谱特征性强、灵敏度高、制样简单,以及检测快捷等优点【10~15】。但是,样本的SERS信号强度不但取决于待测样本中待测物质的浓度,而且与SERS增强基底物理性质(如纳米银或金胶体的形状、粒径以及聚集度等)有关。而常用SERS增强基底银(金)纳米溶胶的可重现性和稳定性均较差,严重影响样本SERS信号的重现性,导致SERS定量分析结果的精确度远远不到实际定量分析的要求。最近,本研究小组发展了一个适用于表面增强拉曼光谱定量分析的乘子效应模型(Multiplicative effects model for surface-enhanced raman spectroscopy, MEMSERS)【16,17】。 MEMSERS模型能够有效消除SERS增强基底物理性质变化对SERS定量分析结果准确度的影响。本研究将SERS技术与MEMSERS相结合,实现了血浆和药片中甲巯咪唑含量的快速准确定量分析。
2 实验部分
2.1 仪器与试剂
便携式i-拉曼785H光谱仪(上海必达泰克光电科技有限公司);1290液相色谱/G6460B系列三级四极杆质谱联用仪(安捷伦科技有限公司);C18反相色谱柱(150 mm×2.1 mm, 3.5 μm)。
二水合柠檬酸三钠(C6H5Na3O7·2H2O)和AgNO3(Sigma-Aldrich 试剂有限公司)。正常人血浆从双流正龙生化制品研究室(长沙)获得。甲巯咪唑和2-巯基异烟酸(阿拉丁公司)。甲巯咪唑药片(北京燕京药业有限公司)。实验用水均为超纯水(18.2 MΩ cm),直接从艾科浦纯水系统(艾科浦公司)中取用。所有试剂均为分析纯。
2.2 SERS增强基底银纳米颗粒的制备
实验中所用玻璃器皿均在王水(HCl-HNO3, 3∶1, V/V)中浸泡8 h以上,用超纯水冲洗,烘干后使用。本研究中所用银纳米溶胶是按照Lee-Meisel方法【18】制备:称取18 mg AgNO3, 用水溶解并定容至100 mL;将此溶液转至圆底烧瓶并加热至沸腾后,迅速加入2 mL柠檬酸三钠溶液(1%),保持沸腾并继续回流1 h后停止加热,使溶液自然冷却至室温,装瓶待用。
2.3 样品的制备
分别用超纯水和乙醇溶解适量的甲巯咪唑和2-巯基异烟酸,得到5.50 mmol/L甲巯咪唑储备液和0.31 mmol/L 2-巯基异烟酸储备液,于4 ℃保存备用。移取10 μL 2-巯基异烟酸储备液和不同体积的甲巯咪唑储备液,用水稀释至0.50 mL,得到11个甲巯咪唑校正样本,其中甲巯咪唑的浓度分别为0.06, 0.18, 0.37, 0.55, 0.73, 0.92, 1.10, 1.28, 1.47, 1.65和1.83 μmol/L。
移取1.00 mL正常人血浆(用LC-MS/MS没有检测出甲巯咪唑)于10.0 mL离心管中,用乙腈稀释至8.0 mL,于20 ℃以10000 r/min离心20min; 吸取4.00 mL 上清液,定容至50.0 mL,备用。移取10 μL 2-巯基异烟酸储备液与适量甲巯咪唑储备液,用上述血浆上清液稀释至0.5 mL,得到甲巯咪唑浓度分别为0.28, 0.46, 0.83, 1.01, 1.19, 1.38和1.74 μmol/L的血浆预测样本(对每个甲巯咪唑浓度水平,均配制3个重复血浆样本)。
准确称取一片甲巯咪唑药片(0.0721 g),用水溶解并定容至50.0 mL,再以水将其稀释50倍,得到甲巯咪唑药片储备液。如表1所示,将20 μL药片储备液与10 μL 2-巯基异烟酸储备液和适量甲巯咪唑储备液混合后用超纯水稀释至0.5 mL,得到药片预测集样本。
2.4 样本SERS光谱的采集
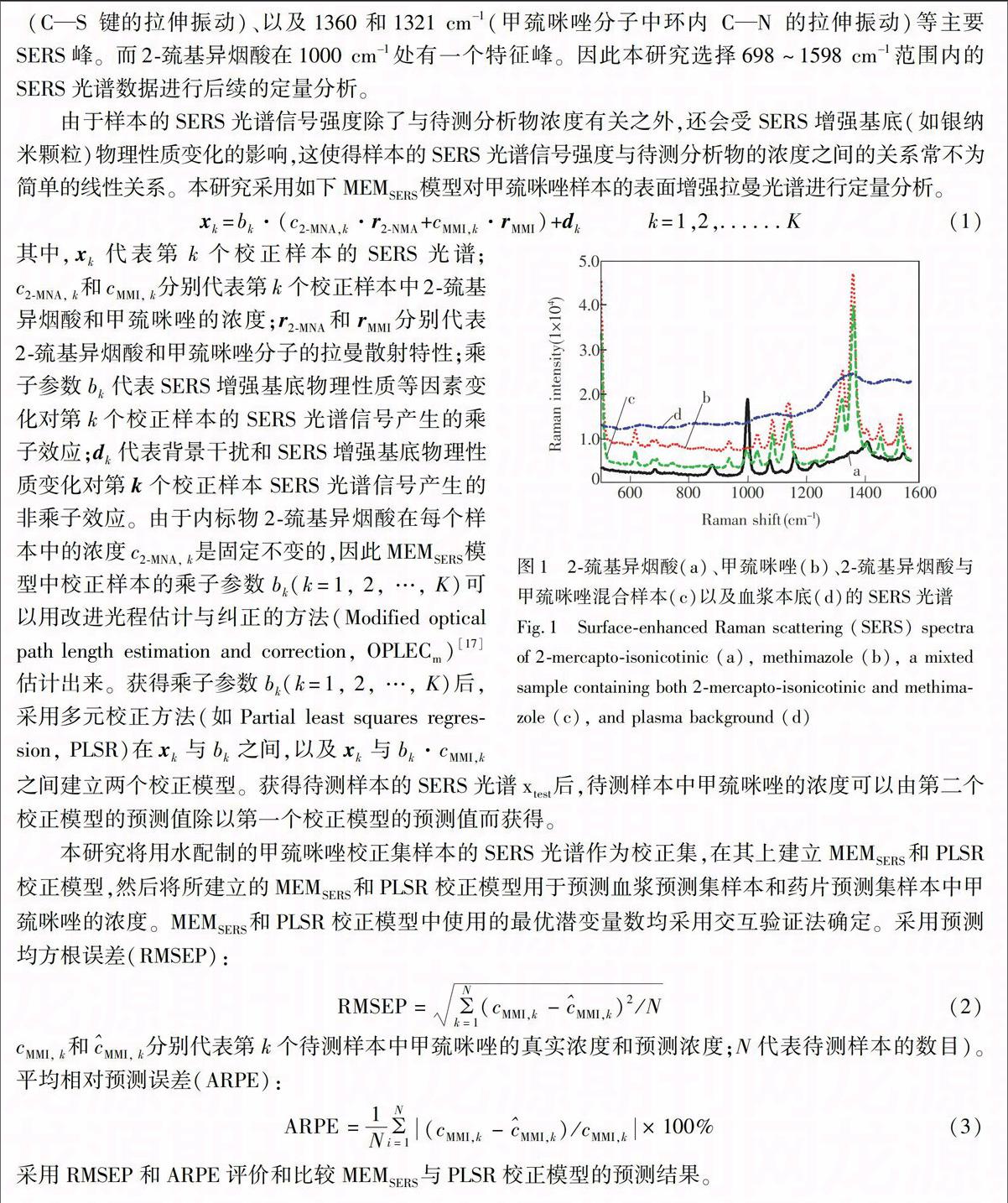
将10 μL样本、50 μL的银纳米颗粒(AgNPs)、以及2.5 μL 2 mol/L KCl混匀后,立即用毛细管取样,使用耦合了BAC151A拉曼影像显微镜采样系统的便携式i-Raman 785H光谱仪采集其SERS光谱。样本SERS光谱采集参数如下:拉曼位移范围500~1600 cm
激光波长785 nm,激光功率300 mW,物镜倍数20倍,曝光时间5 s。每个样品均重复测量3次。
2.5 LC-MS/MS实验
为了验证SERS 技术结合MEMSERS模型对药片预测集样本中甲巯咪唑含量预测结果的准确性,本研究采用1290液相色谱/G6460B系列三级四极杆质谱联用仪对药片预测集中的9个样本中甲巯咪唑的含量进行了检测。每个样本均重复检测3次。具体的色谱与质谱实验条件如下: C18反相色谱柱(150 mm×2.1 mm, 3.5 μm),流动相为0.1%甲酸-甲醇(1∶1,V/V),流速0.3 mL/min,进样量20 μL,质谱离子源:电喷雾离子源,检测模式:正离子多重反应监测模式,母离子:m/z 115.0 (其最优电压值为102 V),子离子:m/z 57.2和88.0 (其最优的碰撞电压分别为18和15 V),扫描时间:300 ms。表1 药片预测样本的实验设计范围内的SERS光谱数据进行后续的定量分析。
由于样本的SERS光谱信号强度除了与待测分析物浓度有关之外,还会受SERS增强基底(如银纳米颗粒)物理性质变化的影响,这使得样本的SERS光谱信号强度与待测分析物的浓度之间的关系常不为简单的线性关系。本研究采用如下MEMSERS模型对甲巯咪唑样本的表面增强拉曼光谱进行定量分析。
xk=bk·(c2-MNA,k·r2-NMA+cMMI,k·rMMI)+dk k=1,2,......K(1)
其中,xk代表第k个校正样本的SERS光谱;c2-MNA, k和cMMI, k分别代表第k个校正样本中2-巯基异烟酸和甲巯咪唑的浓度;r2-MNA和rMMI分别代表2-巯基异烟酸和甲巯咪唑分子的拉曼散射特性;乘子参数bk代表SERS增强基底物理性质等因素变化对第k个校正样本的SERS光谱信号产生的乘子效应;dk代表背景干扰和SERS增强基底物理性质变化对第k个校正样本SERS光谱信号产生的非乘子效应。由于内标物2-巯基异烟酸在每个样本中的浓度c2-MNA, k是固定不变的,因此MEMSERS模型中校正样本的乘子参数bk(k=1, 2, …, K)可以用改进光程估计与纠正的方法(Modified optical path length estimation and correction, OPLECm)【17】估计出来。获得乘子参数bk(k=1, 2, …, K)后,采用多元校正方法(如Partial least squares regression, PLSR)在xk与bk之间,以及xk与bk·cMMI,k之间建立两个校正模型。获得待测样本的SERS光谱xtest后,待测样本中甲巯咪唑的浓度可以由第二个校正模型的预测值除以第一个校正模型的预测值而获得。
本研究将用水配制的甲巯咪唑校正集样本的SERS光谱作为校正集,在其上建立MEMSERS和PLSR校正模型,然后将所建立的MEMSERS和PLSR校正模型用于预测血浆预测集样本和药片预测集样本中甲巯咪唑的浓度。MEMSERS和PLSR校正模型中使用的最优潜变量数均采用交互验证法确定。采用预测均方根误差(RMSEP):
RMSEP=Nk=1(cMMI,k-MMI,k)2/N(2)
cMMI, k和MMI, k分别代表第k个待测样本中甲巯咪唑的真实浓度和预测浓度;N代表待测样本的数目)。平均相对预测误差(ARPE):
ARPE=1NNi=1(cMMI,k-MMI,k)/cMMI,k×100%(3)
采用RMSEP和ARPE评价和比较MEMSERS与PLSR校正模型的预测结果。
3 结果与讨论
3.1 内标浓度的优化
采用内标加入法对待测样本中的甲巯咪唑进行SERS定量分析。内标物2-巯基异烟酸(其SERS特征峰数量少且信号较强)和待测分析物2-巯基异烟酸均含有巯基,它们可通过与银纳米颗粒表面形成稳定的AgS键而修饰在银纳米颗粒表面。由于甲巯咪唑与2-巯基异烟酸之间可能存在对银纳米颗粒表面上结合位点的竞争现象,作为内标物加入的2-巯基异烟酸的浓度若太低,则其SERS信号太弱,起不到内标作用;但2-巯基异烟酸浓度也不能太高,否则会影响甲巯咪唑的检测灵敏度。为了实现对本研究所考察浓度范围内的甲巯咪唑进行较准确的定量分析,将所加入的2-巯基异烟酸的浓度设置为一个比较适中的值,即6.2 μmol/L。
3.2 增强基底物理物质对样本SERS光谱信号的影响
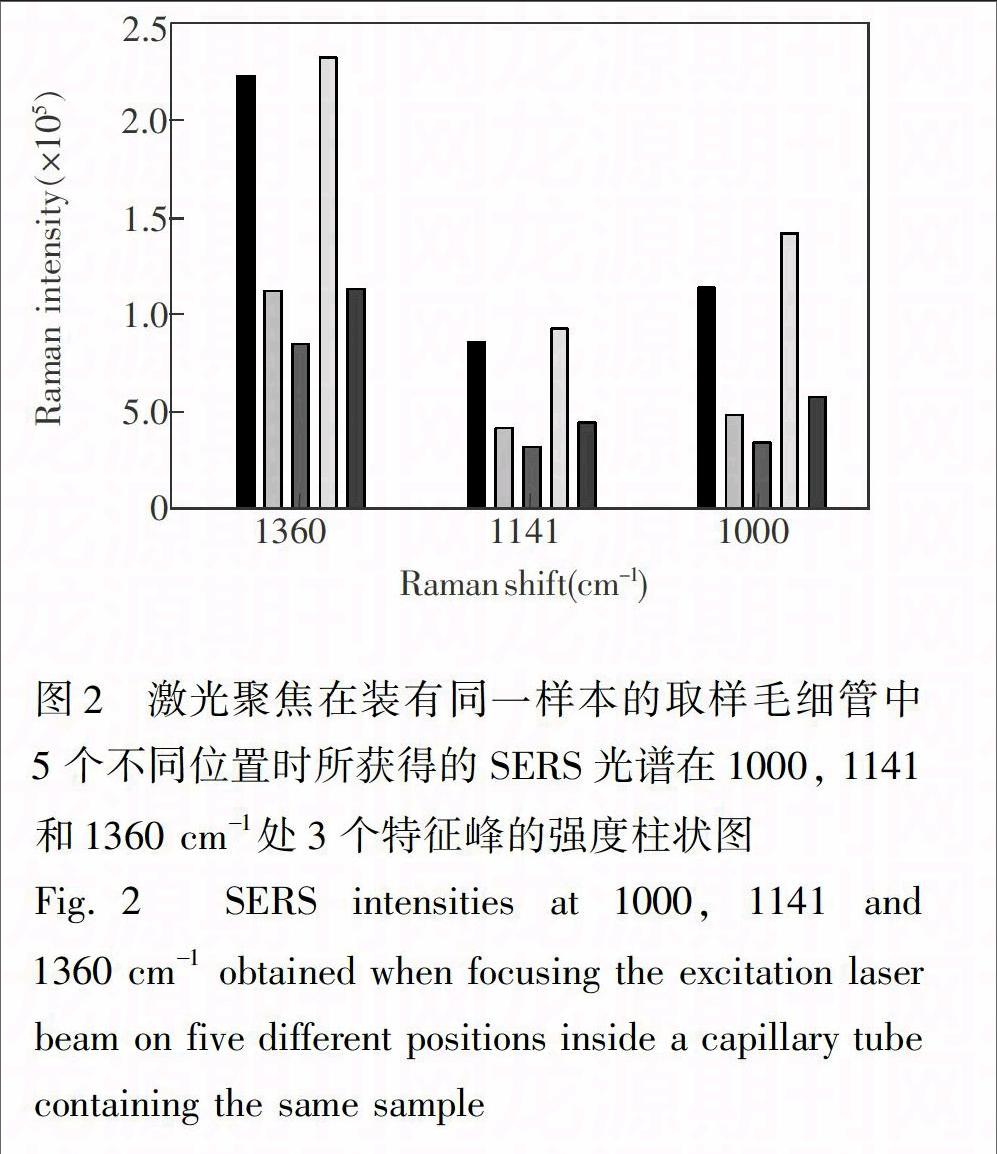
在SERS定量分析过程中遇到的最大问题是:样本的SERS光谱信号重现性较差。如图2所示,在其它实验条件完全一样的情况下,将激光聚焦在同一根取样毛细管(毛细管中装有同一银纳米颗粒-KCl-样本混合物)中不同位置所获得一组SERS光谱之间存在明显差异。这主要是由SERS增强基底纳米银颗粒的形状和大小分布不均匀所致。由OPLECm所估计出来的甲巯咪唑校正集样本的乘子参数bk在1.0~2.6范围内变动,表明SERS增强基底纳米银颗粒的形状和大小分布不均匀性对样本SERS光谱信号有很大影响。必须采取措施消除这种不利影响,才能实现对血浆预测集样本和药片预测集样本中甲巯咪唑的准确定量分析。
3.3 样品分析
3.3.1 血浆样本中甲巯咪唑的定量分析 将建立在用水配制的甲巯咪唑校正集样本SERS光谱数据上的MEMSERS和PLSR校正模型应用于血浆预测集样本中甲巯咪唑浓度的定量分析。如表2所示,PLSR模型的RMSEP和ARPE值分别为0.13 μmol/L和11.0%。PLSR模型预测值与实际值之间的偏差相对较大。此外,PLSR模型对同一样本的3次重复测量SERS光谱的定量分析结果的标准方差较大。这些现象表明, PLSR模型不能有效消除SERS增强基底物理性质的不均一性对SERS定量分析结果的不利影响。相比之下,MEMSERS模型给出的甲巯咪唑浓度的预测值与实际值非常接近,而且其对同一样本的3次重复测量SERS光谱的定量分析结果的标准方差也较小。MEMSERS的RMSEP和ARPE值分别为0.05 μmol/L和5.1%,均明显小于PLSR模型的相应值。另外,通过估算得到MEMSERS对血浆体系中甲巯咪唑的检出限为32 nmol/L,优于文献中采用的气相色谱-质谱法的检出限(45 nmol/L)【5, 6】。
为了验证MEMSERS模型预测结果的可靠性,采用另一份正常人的血浆(新血浆),按照上述血浆预测样本的配制方法重新配制一组血浆预测样本,并使用建立的MEMSERS模型对这一组血浆预测样本中的甲巯咪唑浓度进行定量分析。结果表明,MEMSERS模型的预测结果的准确度很高,其RMSEP和ARPE值分别为0.06 μmol/L和5.4%,与前面所获得结果几乎完全一致。因此,待测样本基质的变化也不影响MEMSERS对血浆预测集样本中甲巯咪唑的定量分析结果。
3.3.2 药片中甲巯咪唑的定量分析 SERS技术结合MEMSERS模型和LC-MS/MS对一片药片中甲巯咪唑含量的测定值分别为5.4和5.0 mg。这两种方法的检测结果基本一致,并且十分接近该药片所附说明书上标注的规格(5 mg/片)。表3列出了MEMSERS和 LC-MS/MS对药片预测集样本中甲巯咪唑的定量分析结果。从表3可知,MEMSERS定量分析结果的平均加标回收率在93.3%~110.9%之间,与LC-MS/MS定量分析结果的准确度基本一致,表明SERS技术结合MEMSERS模型方法能准确测定复杂体系中甲巯咪唑的含量,并且有望发展成为复杂体系中甲巯咪唑含量的常规定量分析方法。表3 MEMSERS和 LC-MS/MS对药片预测样本中甲巯咪唑的定量分析结果
4 结 论
采用了MEMSERS模型消除SERS增强基底物理性质的不均一性对SERS定量分析结果的不利影响,以实现血浆和药片样本中甲巯咪唑的准确定量检测。结果表明,建立在超纯水配制的甲巯咪唑校正集样本SERS光谱数据上的MEMSERS模型能够从血浆预测集样本和药片预测集样本的SERS光谱中准确预测出相应甲巯咪唑的含量。MEMSERS对血浆预测集样本中甲巯咪唑含量的平均相对预测误差约为5.1%,检出限达到了32 nmol/L,而且待测样本的基质变化不影响MEMSERS定量分析结果的准确度;MEMSERS模型对药片预测集样本中甲巯咪唑含量的预测结果的回收率在93.3%~110.9%之间,与LC-MS/MS对照实验的结果基本一致。SERS技术与MEMSERS模型相结合检测甲巯咪唑的方法具有简便、快捷、灵敏度高、检出限低的特点,有望发展成为复杂体系中甲巯咪唑含量的常规定量分析方法。
References
1 Weetman A P, McGregor A M, Hall R. Clin. Endocrinol., 1984, 21(2): 163-172
2 Kendall-Taylor P. Br. Med. J. 1984, 288(6416): 509
3 Hollosi L, Kettrup A, Schramm K W. J. Pharm. Biom. Anal., 2004, 36(4): 921-924
4 Kusmierek K, Bald E. Talanta, 2007, 71(5): 2121-2125
5 Zou Q H, Liu Y, Xie M X, Han J, Zhang L. Anal. Chim. Acta, 2005, 551(1): 184-191
6 Zhang L, Liu Y, Xie M X, Qiu Y M. J. Chromatogr. A, 2005, 1074(1): 1-7
7 Shahrokhian S, Ghalkhani M. Electroanalysis, 2008, 20(10): 1061-1066
8 Sun J Y, Zhang C Y, Xiao X L, Niu L, You T Y, Wang E K. Electroanalysis, 2005, 17(18): 1675-1680
9 Economou A, Tzanavaras P D, Notou M, Themelis D G. Anal. Chim. Acta, 2004, 505(1): 129-133
10 Kneipp K, Wang Y, Kneipp H, Perelman L T, Itzkan I, Dasari R R, Feld M S. Phys. Rev. Lett., 1997, 78(9): 1667
11 Nie S, Emory S R. Science, 1997, 275(5303): 1102-1106
12 LIANG Man-Fen, ZHOU Dian-Ming, WANG Yu, CHEN Cui-Hua, JIANG Jian-Hui. Chinese J. Anal. Chem., 2013, 41(9): 1341-1347
梁满芬, 周殿明, 王 玉, 陈翠花, 蒋健晖. 分析化学, 2013, 41(9): 1341-1347
13 Otto A, Mrozek I, Grabhorn H, Akemann W. J. Phys.: Condens. Matter, 1992, 4(5): 1143
14 NI Dan-Dan, WANG Wei-Wei, YAO Jian-Lin, ZHANG Xie-Jiao, GU Ren-Ao. Spectroscopy and Spectral Analysis, 2011, 31(2): 394-397
倪丹丹, 王伟伟, 姚建林, 张雪娇, 顾仁敖. 光谱学与光谱分析, 2011, 31(2): 394-397
15 FAN Xiao-Min, ZOU Wen-Jun, GU Ren-Ao, YAO Jina-Lin. Chem. J. Chinese Universities, 2008, 29(1): 130-134
范晓敏, 邹文君, 顾仁敖, 姚建林. 高等学校化学学报, 2008, 29(1): 130-134
16 Xia T H, Chen Z P, Chen Y, Tin J W, Yu R Q. Anal. Methods, 2014, 6(7): 2363-2370
17 Song J, Chen Z P, Jin J W, Chen Y, Yu R Q. Chemom. Intell. Lab. Syst., 2014, 135: 31-36
18 Lee P C, Meisel D. J. Phys. Chem., 1982, 86(17): 3391-3395
19 Tin J W, Chen Z P, Li L M, Steponavicius R, Thennadil S N, Yang J, Yu R Q. Anal. Chem., 2011, 84(1): 320-326
摘 要 将表面增强拉曼散射(Surface-enhanced raman scattering,SERS)技术与乘子效应模型结合,采用2-巯基异烟酸作为内标,以银纳米颗粒为SERS增强基底,对血浆和药片样本中甲巯咪唑的进行准确的定量分析。实验结果表明,本方法对血浆样本中甲巯咪唑的平均相对预测误差为5.1%,检出限为32 nmol/L;对药片中甲巯咪唑的定量结果与LC-MS/MS方法基本一致,其加标回收率在93.3%~110.9%之间。
关键词 表面增强拉曼光谱; 甲巯咪唑; 2-巯基异烟酸; 乘子效应模型; 定量分析
1 引 言
甲巯咪唑为抗甲状腺药物,一般用于各种类型的甲状腺功能亢进症【1,2】的治疗。但该药物的使用不当或者过量使用会产生一定的副作用,轻则出现粒细胞减少,脱发、皮炎等,重则可能导致中毒性肝损伤。因此,必须在甲巯咪唑的生产和使用过程进行严格的定量监控。目前,甲巯咪唑的定量分析方法有液相色谱法【3,4】、气相色谱-质谱法【5,6】、电化学方法【7,8】和间接流动注射法【9】等。但是,这些方法均具有一定的局限性:需要对复杂实际样品进行繁琐费时的预处理,或者所需仪器价格昂贵且体积庞大,不适用于大量样品的快速在线分析。因此, 建立一种新型简便、快速、灵敏的甲巯咪唑的定量方法仍具有较重要的现实意义。
表面增强拉曼散射技术(Surface-enhanced raman scattering,SERS)具有光谱特征性强、灵敏度高、制样简单,以及检测快捷等优点【10~15】。但是,样本的SERS信号强度不但取决于待测样本中待测物质的浓度,而且与SERS增强基底物理性质(如纳米银或金胶体的形状、粒径以及聚集度等)有关。而常用SERS增强基底银(金)纳米溶胶的可重现性和稳定性均较差,严重影响样本SERS信号的重现性,导致SERS定量分析结果的精确度远远不到实际定量分析的要求。最近,本研究小组发展了一个适用于表面增强拉曼光谱定量分析的乘子效应模型(Multiplicative effects model for surface-enhanced raman spectroscopy, MEMSERS)【16,17】。 MEMSERS模型能够有效消除SERS增强基底物理性质变化对SERS定量分析结果准确度的影响。本研究将SERS技术与MEMSERS相结合,实现了血浆和药片中甲巯咪唑含量的快速准确定量分析。
2 实验部分
2.1 仪器与试剂
便携式i-拉曼785H光谱仪(上海必达泰克光电科技有限公司);1290液相色谱/G6460B系列三级四极杆质谱联用仪(安捷伦科技有限公司);C18反相色谱柱(150 mm×2.1 mm, 3.5 μm)。
二水合柠檬酸三钠(C6H5Na3O7·2H2O)和AgNO3(Sigma-Aldrich 试剂有限公司)。正常人血浆从双流正龙生化制品研究室(长沙)获得。甲巯咪唑和2-巯基异烟酸(阿拉丁公司)。甲巯咪唑药片(北京燕京药业有限公司)。实验用水均为超纯水(18.2 MΩ cm),直接从艾科浦纯水系统(艾科浦公司)中取用。所有试剂均为分析纯。
2.2 SERS增强基底银纳米颗粒的制备
实验中所用玻璃器皿均在王水(HCl-HNO3, 3∶1, V/V)中浸泡8 h以上,用超纯水冲洗,烘干后使用。本研究中所用银纳米溶胶是按照Lee-Meisel方法【18】制备:称取18 mg AgNO3, 用水溶解并定容至100 mL;将此溶液转至圆底烧瓶并加热至沸腾后,迅速加入2 mL柠檬酸三钠溶液(1%),保持沸腾并继续回流1 h后停止加热,使溶液自然冷却至室温,装瓶待用。
2.3 样品的制备
分别用超纯水和乙醇溶解适量的甲巯咪唑和2-巯基异烟酸,得到5.50 mmol/L甲巯咪唑储备液和0.31 mmol/L 2-巯基异烟酸储备液,于4 ℃保存备用。移取10 μL 2-巯基异烟酸储备液和不同体积的甲巯咪唑储备液,用水稀释至0.50 mL,得到11个甲巯咪唑校正样本,其中甲巯咪唑的浓度分别为0.06, 0.18, 0.37, 0.55, 0.73, 0.92, 1.10, 1.28, 1.47, 1.65和1.83 μmol/L。
移取1.00 mL正常人血浆(用LC-MS/MS没有检测出甲巯咪唑)于10.0 mL离心管中,用乙腈稀释至8.0 mL,于20 ℃以10000 r/min离心20min; 吸取4.00 mL 上清液,定容至50.0 mL,备用。移取10 μL 2-巯基异烟酸储备液与适量甲巯咪唑储备液,用上述血浆上清液稀释至0.5 mL,得到甲巯咪唑浓度分别为0.28, 0.46, 0.83, 1.01, 1.19, 1.38和1.74 μmol/L的血浆预测样本(对每个甲巯咪唑浓度水平,均配制3个重复血浆样本)。
准确称取一片甲巯咪唑药片(0.0721 g),用水溶解并定容至50.0 mL,再以水将其稀释50倍,得到甲巯咪唑药片储备液。如表1所示,将20 μL药片储备液与10 μL 2-巯基异烟酸储备液和适量甲巯咪唑储备液混合后用超纯水稀释至0.5 mL,得到药片预测集样本。
2.4 样本SERS光谱的采集
将10 μL样本、50 μL的银纳米颗粒(AgNPs)、以及2.5 μL 2 mol/L KCl混匀后,立即用毛细管取样,使用耦合了BAC151A拉曼影像显微镜采样系统的便携式i-Raman 785H光谱仪采集其SERS光谱。样本SERS光谱采集参数如下:拉曼位移范围500~1600 cm
激光波长785 nm,激光功率300 mW,物镜倍数20倍,曝光时间5 s。每个样品均重复测量3次。
2.5 LC-MS/MS实验
为了验证SERS 技术结合MEMSERS模型对药片预测集样本中甲巯咪唑含量预测结果的准确性,本研究采用1290液相色谱/G6460B系列三级四极杆质谱联用仪对药片预测集中的9个样本中甲巯咪唑的含量进行了检测。每个样本均重复检测3次。具体的色谱与质谱实验条件如下: C18反相色谱柱(150 mm×2.1 mm, 3.5 μm),流动相为0.1%甲酸-甲醇(1∶1,V/V),流速0.3 mL/min,进样量20 μL,质谱离子源:电喷雾离子源,检测模式:正离子多重反应监测模式,母离子:m/z 115.0 (其最优电压值为102 V),子离子:m/z 57.2和88.0 (其最优的碰撞电压分别为18和15 V),扫描时间:300 ms。表1 药片预测样本的实验设计范围内的SERS光谱数据进行后续的定量分析。
由于样本的SERS光谱信号强度除了与待测分析物浓度有关之外,还会受SERS增强基底(如银纳米颗粒)物理性质变化的影响,这使得样本的SERS光谱信号强度与待测分析物的浓度之间的关系常不为简单的线性关系。本研究采用如下MEMSERS模型对甲巯咪唑样本的表面增强拉曼光谱进行定量分析。
xk=bk·(c2-MNA,k·r2-NMA+cMMI,k·rMMI)+dk k=1,2,......K(1)
其中,xk代表第k个校正样本的SERS光谱;c2-MNA, k和cMMI, k分别代表第k个校正样本中2-巯基异烟酸和甲巯咪唑的浓度;r2-MNA和rMMI分别代表2-巯基异烟酸和甲巯咪唑分子的拉曼散射特性;乘子参数bk代表SERS增强基底物理性质等因素变化对第k个校正样本的SERS光谱信号产生的乘子效应;dk代表背景干扰和SERS增强基底物理性质变化对第k个校正样本SERS光谱信号产生的非乘子效应。由于内标物2-巯基异烟酸在每个样本中的浓度c2-MNA, k是固定不变的,因此MEMSERS模型中校正样本的乘子参数bk(k=1, 2, …, K)可以用改进光程估计与纠正的方法(Modified optical path length estimation and correction, OPLECm)【17】估计出来。获得乘子参数bk(k=1, 2, …, K)后,采用多元校正方法(如Partial least squares regression, PLSR)在xk与bk之间,以及xk与bk·cMMI,k之间建立两个校正模型。获得待测样本的SERS光谱xtest后,待测样本中甲巯咪唑的浓度可以由第二个校正模型的预测值除以第一个校正模型的预测值而获得。
本研究将用水配制的甲巯咪唑校正集样本的SERS光谱作为校正集,在其上建立MEMSERS和PLSR校正模型,然后将所建立的MEMSERS和PLSR校正模型用于预测血浆预测集样本和药片预测集样本中甲巯咪唑的浓度。MEMSERS和PLSR校正模型中使用的最优潜变量数均采用交互验证法确定。采用预测均方根误差(RMSEP):
RMSEP=Nk=1(cMMI,k-MMI,k)2/N(2)
cMMI, k和MMI, k分别代表第k个待测样本中甲巯咪唑的真实浓度和预测浓度;N代表待测样本的数目)。平均相对预测误差(ARPE):
ARPE=1NNi=1(cMMI,k-MMI,k)/cMMI,k×100%(3)
采用RMSEP和ARPE评价和比较MEMSERS与PLSR校正模型的预测结果。
3 结果与讨论
3.1 内标浓度的优化
采用内标加入法对待测样本中的甲巯咪唑进行SERS定量分析。内标物2-巯基异烟酸(其SERS特征峰数量少且信号较强)和待测分析物2-巯基异烟酸均含有巯基,它们可通过与银纳米颗粒表面形成稳定的AgS键而修饰在银纳米颗粒表面。由于甲巯咪唑与2-巯基异烟酸之间可能存在对银纳米颗粒表面上结合位点的竞争现象,作为内标物加入的2-巯基异烟酸的浓度若太低,则其SERS信号太弱,起不到内标作用;但2-巯基异烟酸浓度也不能太高,否则会影响甲巯咪唑的检测灵敏度。为了实现对本研究所考察浓度范围内的甲巯咪唑进行较准确的定量分析,将所加入的2-巯基异烟酸的浓度设置为一个比较适中的值,即6.2 μmol/L。
3.2 增强基底物理物质对样本SERS光谱信号的影响
在SERS定量分析过程中遇到的最大问题是:样本的SERS光谱信号重现性较差。如图2所示,在其它实验条件完全一样的情况下,将激光聚焦在同一根取样毛细管(毛细管中装有同一银纳米颗粒-KCl-样本混合物)中不同位置所获得一组SERS光谱之间存在明显差异。这主要是由SERS增强基底纳米银颗粒的形状和大小分布不均匀所致。由OPLECm所估计出来的甲巯咪唑校正集样本的乘子参数bk在1.0~2.6范围内变动,表明SERS增强基底纳米银颗粒的形状和大小分布不均匀性对样本SERS光谱信号有很大影响。必须采取措施消除这种不利影响,才能实现对血浆预测集样本和药片预测集样本中甲巯咪唑的准确定量分析。
3.3 样品分析
3.3.1 血浆样本中甲巯咪唑的定量分析 将建立在用水配制的甲巯咪唑校正集样本SERS光谱数据上的MEMSERS和PLSR校正模型应用于血浆预测集样本中甲巯咪唑浓度的定量分析。如表2所示,PLSR模型的RMSEP和ARPE值分别为0.13 μmol/L和11.0%。PLSR模型预测值与实际值之间的偏差相对较大。此外,PLSR模型对同一样本的3次重复测量SERS光谱的定量分析结果的标准方差较大。这些现象表明, PLSR模型不能有效消除SERS增强基底物理性质的不均一性对SERS定量分析结果的不利影响。相比之下,MEMSERS模型给出的甲巯咪唑浓度的预测值与实际值非常接近,而且其对同一样本的3次重复测量SERS光谱的定量分析结果的标准方差也较小。MEMSERS的RMSEP和ARPE值分别为0.05 μmol/L和5.1%,均明显小于PLSR模型的相应值。另外,通过估算得到MEMSERS对血浆体系中甲巯咪唑的检出限为32 nmol/L,优于文献中采用的气相色谱-质谱法的检出限(45 nmol/L)【5, 6】。
为了验证MEMSERS模型预测结果的可靠性,采用另一份正常人的血浆(新血浆),按照上述血浆预测样本的配制方法重新配制一组血浆预测样本,并使用建立的MEMSERS模型对这一组血浆预测样本中的甲巯咪唑浓度进行定量分析。结果表明,MEMSERS模型的预测结果的准确度很高,其RMSEP和ARPE值分别为0.06 μmol/L和5.4%,与前面所获得结果几乎完全一致。因此,待测样本基质的变化也不影响MEMSERS对血浆预测集样本中甲巯咪唑的定量分析结果。
3.3.2 药片中甲巯咪唑的定量分析 SERS技术结合MEMSERS模型和LC-MS/MS对一片药片中甲巯咪唑含量的测定值分别为5.4和5.0 mg。这两种方法的检测结果基本一致,并且十分接近该药片所附说明书上标注的规格(5 mg/片)。表3列出了MEMSERS和 LC-MS/MS对药片预测集样本中甲巯咪唑的定量分析结果。从表3可知,MEMSERS定量分析结果的平均加标回收率在93.3%~110.9%之间,与LC-MS/MS定量分析结果的准确度基本一致,表明SERS技术结合MEMSERS模型方法能准确测定复杂体系中甲巯咪唑的含量,并且有望发展成为复杂体系中甲巯咪唑含量的常规定量分析方法。表3 MEMSERS和 LC-MS/MS对药片预测样本中甲巯咪唑的定量分析结果
4 结 论
采用了MEMSERS模型消除SERS增强基底物理性质的不均一性对SERS定量分析结果的不利影响,以实现血浆和药片样本中甲巯咪唑的准确定量检测。结果表明,建立在超纯水配制的甲巯咪唑校正集样本SERS光谱数据上的MEMSERS模型能够从血浆预测集样本和药片预测集样本的SERS光谱中准确预测出相应甲巯咪唑的含量。MEMSERS对血浆预测集样本中甲巯咪唑含量的平均相对预测误差约为5.1%,检出限达到了32 nmol/L,而且待测样本的基质变化不影响MEMSERS定量分析结果的准确度;MEMSERS模型对药片预测集样本中甲巯咪唑含量的预测结果的回收率在93.3%~110.9%之间,与LC-MS/MS对照实验的结果基本一致。SERS技术与MEMSERS模型相结合检测甲巯咪唑的方法具有简便、快捷、灵敏度高、检出限低的特点,有望发展成为复杂体系中甲巯咪唑含量的常规定量分析方法。
References
1 Weetman A P, McGregor A M, Hall R. Clin. Endocrinol., 1984, 21(2): 163-172
2 Kendall-Taylor P. Br. Med. J. 1984, 288(6416): 509
3 Hollosi L, Kettrup A, Schramm K W. J. Pharm. Biom. Anal., 2004, 36(4): 921-924
4 Kusmierek K, Bald E. Talanta, 2007, 71(5): 2121-2125
5 Zou Q H, Liu Y, Xie M X, Han J, Zhang L. Anal. Chim. Acta, 2005, 551(1): 184-191
6 Zhang L, Liu Y, Xie M X, Qiu Y M. J. Chromatogr. A, 2005, 1074(1): 1-7
7 Shahrokhian S, Ghalkhani M. Electroanalysis, 2008, 20(10): 1061-1066
8 Sun J Y, Zhang C Y, Xiao X L, Niu L, You T Y, Wang E K. Electroanalysis, 2005, 17(18): 1675-1680
9 Economou A, Tzanavaras P D, Notou M, Themelis D G. Anal. Chim. Acta, 2004, 505(1): 129-133
10 Kneipp K, Wang Y, Kneipp H, Perelman L T, Itzkan I, Dasari R R, Feld M S. Phys. Rev. Lett., 1997, 78(9): 1667
11 Nie S, Emory S R. Science, 1997, 275(5303): 1102-1106
12 LIANG Man-Fen, ZHOU Dian-Ming, WANG Yu, CHEN Cui-Hua, JIANG Jian-Hui. Chinese J. Anal. Chem., 2013, 41(9): 1341-1347
梁满芬, 周殿明, 王 玉, 陈翠花, 蒋健晖. 分析化学, 2013, 41(9): 1341-1347
13 Otto A, Mrozek I, Grabhorn H, Akemann W. J. Phys.: Condens. Matter, 1992, 4(5): 1143
14 NI Dan-Dan, WANG Wei-Wei, YAO Jian-Lin, ZHANG Xie-Jiao, GU Ren-Ao. Spectroscopy and Spectral Analysis, 2011, 31(2): 394-397
倪丹丹, 王伟伟, 姚建林, 张雪娇, 顾仁敖. 光谱学与光谱分析, 2011, 31(2): 394-397
15 FAN Xiao-Min, ZOU Wen-Jun, GU Ren-Ao, YAO Jina-Lin. Chem. J. Chinese Universities, 2008, 29(1): 130-134
范晓敏, 邹文君, 顾仁敖, 姚建林. 高等学校化学学报, 2008, 29(1): 130-134
16 Xia T H, Chen Z P, Chen Y, Tin J W, Yu R Q. Anal. Methods, 2014, 6(7): 2363-2370
17 Song J, Chen Z P, Jin J W, Chen Y, Yu R Q. Chemom. Intell. Lab. Syst., 2014, 135: 31-36
18 Lee P C, Meisel D. J. Phys. Chem., 1982, 86(17): 3391-3395
19 Tin J W, Chen Z P, Li L M, Steponavicius R, Thennadil S N, Yang J, Yu R Q. Anal. Chem., 2011, 84(1): 320-326
