摘 要 以纤维素和邻乙酰水杨酰氯为原料, 合成了纤维素邻乙酰水杨酸酯类手性固定相, 考察了其手性拆分能力, 与Chiralcel OJ手性色谱柱进行了比较, 并研究了流动相的组成以及衍生物中双酯羰基的结构对其拆分效果的影响。以红外光谱技术、热重分析等检测手段对获得的纤维素衍生物进行结构表征和分析。在对4种流动相: 正己烷异丙醇、正己烷乙醇、正己烷甲醇异丙醇、正己烷甲醇1,2二氯乙烷进行比较筛选后, 以正己烷异丙醇(90∶10~80∶20, V/V, 0.1% DEA或TFA)体系作流动相, 在正相色谱模式下, 对儿茶酚胺类及酰胺类手性药物进行了手性拆分, 结果表明, 三氟乙酸和二乙胺的最佳使用量均为0.1%, 双酯羰基结构的氢键、偶极作用和苯环的ππ作用相互协同确实有助于该类固定相手性拆分作用的形成。
关键词 ;邻乙酰水杨酸酯; 纤维素; 手性固定相
1 引 言
手性制药一直是医药研究的前沿领域[1], 这是因为消旋体作为药物使用具有一定的风险, 两个对映体通常表现出不同的药效活性, 甚至其中之一还有毒性[2]。因此, 发展高效、快捷的手性药物拆分测定方法对用药安全具有重要的意义[3,4]。
据统计, 有近90%的手性样品可以在纤维素衍生物作为手性固定相上获得拆分[5~7]。Okamoto研究小组[8,9]曾证明纤维素三苯酯中苯基上取代基的类型和位置对纤维素衍生物的光学拆分能力有显著的影响——纤维素衍生物苯环上的取代基通过吸电子或斥电子作用减少或增加苯环和羰基氧上的电子云密度, 进而影响手性识别能力。 目前, 此类苯环取代基大都是简单的烃基或卤原子, 选择范围较窄, 而纤维素作为多羟基化合物, 凡是具有能与羟基键合的官能团的化合物均能对其修饰。受此启发并根据手性拆分的机理, 本研究组认为乙酰水杨酰氯是一种很有潜力的手性拆分用纤维素衍生化试剂, 其邻位的酯基既能提供一个羰基, 又对苯环起到活化并给电子的作用, 如与纤维素成酯, 将同时出现两个活泼酯羰基, 这对于手性拆分是十分有利的。而且目前该类纤维素衍生物无论在纤维素研究中, 还是手性拆分领域均未有见报道。本实验合成了邻乙酰水杨酸酯类手性固定相(Acelylsalicylte chiral stationary phase, ACSP), 针对常用的氯苯那敏、儿茶酚胺类及酰胺类药物进行手性拆分, 根据拆分结果探讨其规律与特点, 并与商品柱进行比较, 研究其优势或不足, 为纤维素类手性固定相的种类扩展提供新的思路。
2 实验部分
2.1 仪器与试剂
Nicolet370FTIR红外光谱仪(美国Thermo公司); TGA热重分析仪(Q500系统, 美国Waters公司); LC30A高效液相色谱工作站(日本岛津公司), 配置SPDM20A二极管阵列检测器; 商品柱Chiralcel OJ(由河南省食品药品检验所提供)。
微晶纤维素(聚合度DP≈100, Merck公司); 邻乙酰水杨酰氯、γ氨丙基三乙氧基硅烷(KH550, 上海阿拉丁试剂公司); 硅胶(粒径为5 μm, 孔径为120 , 日本Daiso公司); 无水N,N二甲基乙酰胺(DMAc)、氯化锂、无水吡啶(分析纯, 天津市科密欧化学试剂有限公司); 甲醇、乙醇、1,2二氯乙烷、异丙醇和正己烷
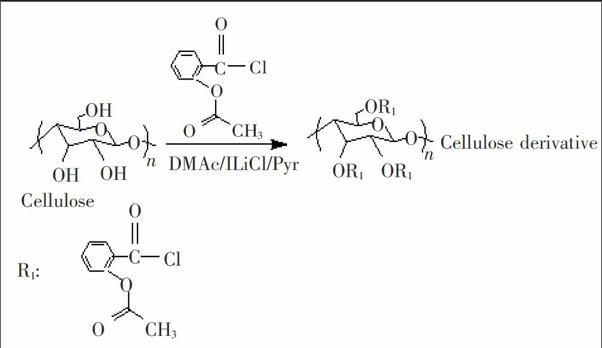
2.2 酯化衍生纤维素的合成
将在80℃真空干燥6 h的纤维素1 g加入70 mL无水DMAc中, 130℃搅拌回流5 h; 冷至室温, 加入3 g 无水LiCl; 搅拌至纤维素完全溶解后[10], 重新升温至100℃, 加入无水吡啶15 mL、7.4 g 邻乙酰水杨酰氯, 反应24 h后, 用大量甲醇沉淀、洗涤、过滤, 40℃真空干燥过夜后, 得纤维素衍生物5.6 g(见图2)。
2.3 氨丙基硅胶的制备
将3 g硅胶与160 mL无水甲苯加入烧瓶中, 氮气保护下加热回流, 除去吸附水。缓缓加入5 mL KH550与1 mL吡啶, 并加热至105℃, 回流。反应完后抽滤, 洗涤, 减压干燥后得氨丙基硅胶[11]。经热重分析, 键合量为7.7%(w/w)。
2.4 HPLC手性固定相的制备
称取1 g纤维素衍生物,溶于40 mL四氢呋喃中, 分次涂敷在3 g氨丙基硅胶表面,并旋转蒸发除去四氢呋喃[12], 如此反复多次后即得ACSP。经热重分析, 涂敷量约为25.1%(w/w)。
2.5 色谱条件
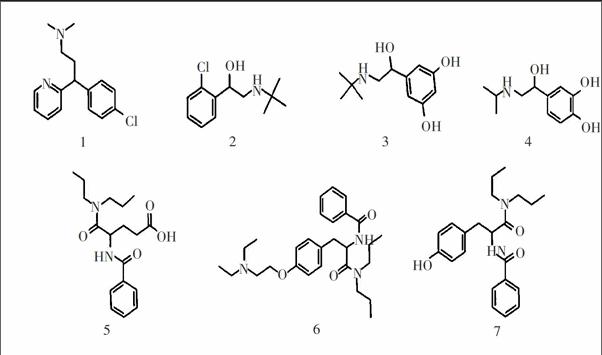
色谱柱(15 cm×0.46 cm i.d.)。以正己烷异丙醇(80∶20, V/V)为匀浆液及填充液, 采用匀浆法装柱。检测条件: 柱温30℃; 检测波长段: 190~400 nm(附图均为各手性药物的最大吸收波长处的色谱图, 无最大吸收者, 以220 nm波长处色谱峰为主); 以正己烷醇为流动相(可添加少量二乙胺(DEA)或三氟乙酸(TFA))。流速为1 mL/min, 以1,3,5三叔丁基苯测定死时间t0, 均以流动相溶解手性化合物, 浓度约为80 μg/mL, 进样量: 20 μL。
固定相的拆分能力测试与考察
对7种手性药物进行拆分评价, 并以下列参数对OJ柱、ACSP进行考察: 保留因子k=(t-t0)/t0, 其中t0为死时间, 由1,3,5三叔丁基苯来测定; t为对映单体的保留时间; 分离因子α=k2/k1; 分离度Rs与理论板数n均由色谱软件自动计算。
3.2.1 与OJ柱的比较及拆分机理 经过色谱条件的优化后, OJ柱、ACSP的最佳拆分条件及结果见表1和图4。表1中, 对于消旋体1、2、3及4, OJ柱分离度明显弱于ACSP, 保留时间也更长, 因此拆分效果不及ACSP。其中儿茶酚胺类消旋体2、3及4的手性中心均与羟基和苯环相连接, ACSP分离度优于OJ柱的原因可能是ACSP中苯环上的双酯基与消旋体2、3及4的手性中心上的羟基发生的氢键作用强于OJ柱上的单酯基。
消旋体2的苯环上有一邻位氯, 对苯环吸电子, 消旋体3的苯环上有两个间位羟基, 对苯环供电子, 而ACSP中的邻位酯基也对苯环供电子, 因此, 消旋体2与ACSP发生的ππ作用强于药物3与ACSP, 从而导致在ACSP的拆分中, 消旋体2的保留时间长于消旋体3, 分离效果也优于消旋体3。消旋体3与4的侧链氨基取代基不同: 消旋体3中为叔丁基, 4中为异丙基。在ACSP上, 消旋体3的保留时间短于4, 但分离效果却优于4, 二者苯环产生ππ作用的能力相近, 应是叔丁基产生的分子位阻大于异丙基, 阻挡了一部分非手性作用力对手性药物的吸附, 因此造成消旋体3的保留时间短, 分离效果好。
消旋体5、6及7为酰胺类化合物, 该类化合物利用其手性中心上酰胺的氢键作用, 能够在刷型固定相上得到有效拆分[13]以及在氨基甲酸酯类纤维素衍生物固定相上得到部分拆分[14], 但在OJ柱上未见拆分迹象, 分析应与OJ固定相上产生氢键作用的官能团较少有关。然而, 消旋体5、6及7却能在ACSP上得到有效分离, 这应归因于三者的酰胺基与水杨酸酯邻位的酯羰基产生的氢键作用和偶极作用。在三者中, 药物5的保留时间最短, 分离效果也最佳: 这是因为其只有一个远离手性中心的苯环(消旋体6和7均有两个), 产生非手性ππ作用力, COOH的电离被三氟乙酸所抑制从而减少了离子作用力, 且分子位阻最小, 能够与ACSP上的手性位点充分接触, 因此能够达到最佳效果。消旋体6中醚取代基产生的分子位阻显然大于7中的羟基, 其分子位阻减少了一些苯环非手性作用力的产生, 因而消旋体6的保留时间短于7, 但由于手性拆分作用力相当, 因此分离因子相差不大。
3.2.2 流动相种类与改性剂对拆分的影响 考察了甲醇、乙醇、1,2二氯乙烷(可少量短时间使用)以及二乙胺、三氟乙酸对固定相拆分能力的影响。选取4号与5号消旋体作为代表, 结果见图5和表2。
由表2可知, 乙醇替换异丙醇后, 洗脱能力得到提高, 但区分能力不及后者。对于消旋体5, 甲醇比例在10%以内时, 随着甲醇用量的提高, 保留时间缩短, 并保证良好的分离效果, 说明甲醇比异丙醇在此固定相上有更佳的洗脱能力与区分能力, 但含量应控制其一定范围内; 而对于保留弱的消旋体4号, 加入甲醇后, 出峰更快, 与固定相的手性作用极不充分, 造成分离不彻底(图5)。二氯乙烷与甲
醇配合使用能够达到极性中和的目的。 从表2可见, 此混合溶剂的洗脱能力对于两类手性药物均弱于异丙醇, 且区分力也不及后者: 消旋体4号只能部分拆分, 且峰形变宽; 消旋体5号未见拆分迹象。分析应是纤维素衍生物已被二氯乙烷部分破坏导致的。综上所述, 4种体系中唯有正己烷异丙醇系统能够对两类消旋体都达到满意的拆分效果, 因此, 本研究以正己烷异丙醇作为流动相。
消旋体5号较特殊, 其不仅具有酰胺结构, 还存在羧基。因此, 考察了流动相正己烷异丙醇(80∶20, V/V)中二乙胺(DEA)和三氟乙酸(TFA)的存在比例对手性拆分的影响, 结果见图6和表3。
由表3可知, 不添加TFA, 消旋体5在45 min内无法洗脱, 这应是羧酸电离后产生的离子作用力导致的;加入0.1% TFA抑制羧酸电离后,即可得到满意的拆分效果。在0.1% TFA存在的情况下, 随着DEA浓度增加, 洗脱时间缩短, 而分离效果基本不变, 但DEA增加至0.3%后, 基线波动较大。羧酸电离的抑制作用直接关系到拆分结果, 加入TFA起关键作用, DEA的存在只起到缩短洗脱时间的作用, 因此, 本研究采用0.1% TFA0.1% DEA为添加剂浓度。4 结 论
本实验通过合成酯取代纤维素衍生物, 将纤维素邻乙酰水杨酸酯用于手性拆分, 取得了良好的拆分效果。初步证明了该纤维素衍生物利用邻位酯羰基的偶极作用和氢键作用配合苯环的ππ作用, 在拆分儿茶酚胺类和手性中心连有酰胺基团的药物方面, 比OJ柱更有优势。充分证明了此衍生物上双酯羰基的强大的手性拆分能力。通过比较流动相发现, 溶剂的种类与相互组合在此手性固定相上表现出的手性区分力差别明显。此外, 根据手性药物酸碱性的差别, 合理添加二乙胺或三氟乙酸等流动相改性剂, 能够达到缩短保留时间、增强拆分效果的作用。
References
1 Ikai T, Okamoto Y. Chem. Rev., 2009, 109(11): 6077-6101
2 Ates H, Mangelings D, Heyden Y V. J. Chromatogr. B, 2008, 875(1): 57-64
3 ZHOU RenDan, LI LaiSheng, CHENG BiaoPing, NIE GuiZhen, ZHANG HongFu. Chinese J. Anal.Chem., 2014, 42(7): 1002-1009
周仁丹, 李来生,程彪平,聂桂珍,张宏福. 分析化学, 2014, 42(7): 1002-1009
4 YAO Na, SONG RuiJuan, FU Yu, SHI HongYu,LONG YuanDe, HUANG TianBao. Chem. J. Chinese Universities, 2008, 29(6): 1102-1106
姚 娜, 宋瑞娟, 富 玉, 石宏宇, 龙远德, 黄天宝. 高等学校化学学报, 2008, 29(6): 1102-1106
5 Tang S W, Li X F, Wang F, Liu G H, Li Y L, Pan F Y. Chirality, 2012, 24(1): 167-173
6 YANG LiPing, WANG LiXin, XU YanLi, QIAN BaoYing, GAO RuYu. Journal of Instrumental Analysis, 2004, 21(5): 25-28
杨丽萍, 王立新, 徐艳丽, 钱宝英, 高如瑜. 分析测试学报,2004, 21(5): 25-28
7 Yamamoto C, Okamoto Y. Bull. Chem. Soc. Jpn, 2004, 77(2): 227-257
8 Okamoto Y, Kauashima M, Hatada K. J. Chromatog., 1986, 363(4): 173-186
9 Okamoto Y, Aburatni R, Hatada K. J. Chromatogr., 1987, 389(1): 95-102
10 Kesavan D, Masakazu H, Jun A, Kousaku O. Cellulose, 2013, 20(1): 365-378
11 Huang J, Chen H, Li T. J. Chromatogr. A, 2006, 1113(12): 109-115
12 QU HaiTao, LI JunQing, SHEN Jun, SHEN XianDe, Yoshio Okamoto. Chinese J. Anal. Chem., 2011, 39(4): 461-465
屈海涛, 李峻青, 沈 军, 沈贤德, 岡本佳男. 分析化学, 2011, 39(4): 461-465
13 LIANG YanMing. Chiral Sepration of Selected Pharmacenticals on rushType Chiral Stationary Phase. Chengdu: Sichuan University, 2003: 36-38
梁彦明. 刷型手性固定相对于药物对映体的拆分研究. 硕士论文. 四川大学, 成都, 2003, 36-38
14 ZHOU Lan, LI GaoLan, YANG GuoSheng. Journal of Shandong University of Science and Technology(Natural Science), 2000, 19(3): 22-24
周 岚, 李高兰, 杨国生. 山东科技大学学报(自然科学版), 2000, 19(3): 22-24
Abstract Cellulose acetylsalicylate chiral stationary phase was synthesized by using cellulose and Oacetylsalicylryl chloride, and evaluated by HPLC. In this work, Chiralcel OJ was also evaluated for comparison. The effects of mobile phase composition and double ester carbonyls of derivative on enantioseparation were investigated. The structure of the obtained derivative was characterized by infrared spectroscopy and thermogravimetric analysis. Hexaneisopropanol (90∶10-80∶20, 0.1% DEA or TFA) was selected by comparison of four mobile phases: namely hexaneisopropanol, hexaneethanol, hexanemethanolisopropanol and hexanemethanoldichloroethane. Seven racemates of catecholamine and amide were used to evaluate its chiral recognition ability in normal phase elution mode and the regularity and characteristics of the novel chiral stationary phase were explored. Based on the chromatographic results, cellulose acetylsalicylate exhibited high enantioseparation ability for catecholamines and some racemates with amide group due to the carbonyls of acetylsalicylate and the optimum amount of DEA or TFA is 0.1%.
Keywords Acetylsalicylate; Cellulose; Chiral stationary phase
(Received 23 January 2015; accepted 23 April 2015)
- 冷酷仙境苯教圣地孜珠寺
- 平民书记白纪年
- “待用公益”席卷全国
- “随手”的力量
- 给孩子加个菜
- 微公益在行动
- 维吉达尼以良心的名义
- 成县秦巴山里的淘宝县
- 冯小燕为了中国好土豆
- 农家电商
- “5100免费水”的水到底有多深?
- 干净是一种力量
- 尧头古镇:沧桑了时光
- 风雨同舟十五年我们一起走过
- 李河君西部掘金成首富
- 欠薪
- 有一种旅行叫探家
- 在中国当农民的韩国女婿
- 海派第一名吃
- 有梦就是未来
- 品茶性达人性
- 谜一样的李蕾
- 李佳“坐火箭”升迁的美女书记
- 中蒙经贸合作与草原丝绸之路经济带构建学术论坛举行
- “十三五”新疆将构建以乌鲁木齐为中心的城际铁路网
- deny yourself
- deodorant
- deodorants
- deodorization
- dep.
- depart
- departable
- depart from sth
- departing
- department
- departmental
- departmentalization
- departmentally
- department of trade and industry
- departmentoftradeandindustry
- departments
- department store
- departmentstore
- department stores
- departs
- departure
- departures
- depend
- dependabilities
- dependability
- 男当下配,女望高门
- 男当家
- 男怕人错行,女怕嫁错郎
- 男怕失足,女怕失身
- 男怕穿靴,女怕戴帽
- 男怕西皮,女怕二簧
- 男怕输笔,女怕输身
- 男性
- 男性佛徒穿的衣
- 男性农民
- 男性和女性
- 男性客人
- 男性家属
- 男性尊长
- 男性巫师
- 男性所恋的女人
- 男性生殖器
- 男性的
- 男性的成年人
- 男性贼人
- 男性长者
- 男性间的同性恋
- 男性间的性行为
- 男愁唱,女愁哭
- 男手