刘春 李嘉驹 代玉兰
摘 要:通过筛选催化剂,改进原料药,合成碘海醇中间体及原料药。目前从磷矿处理液中提取碘项目已取得成功,本方案通过筛选催化剂,改进原料药,合成制备碘海醇中间体及其原料药[1]。
关键词:碘海醇;合成;原料药
中图分类号:TQ421.7 文献标识码:A 文章编号:1671-2064(2020)08-0216-02
1 现有技术路线分析
目前主要形成了三条的技术路线。
1.1 技术路线1
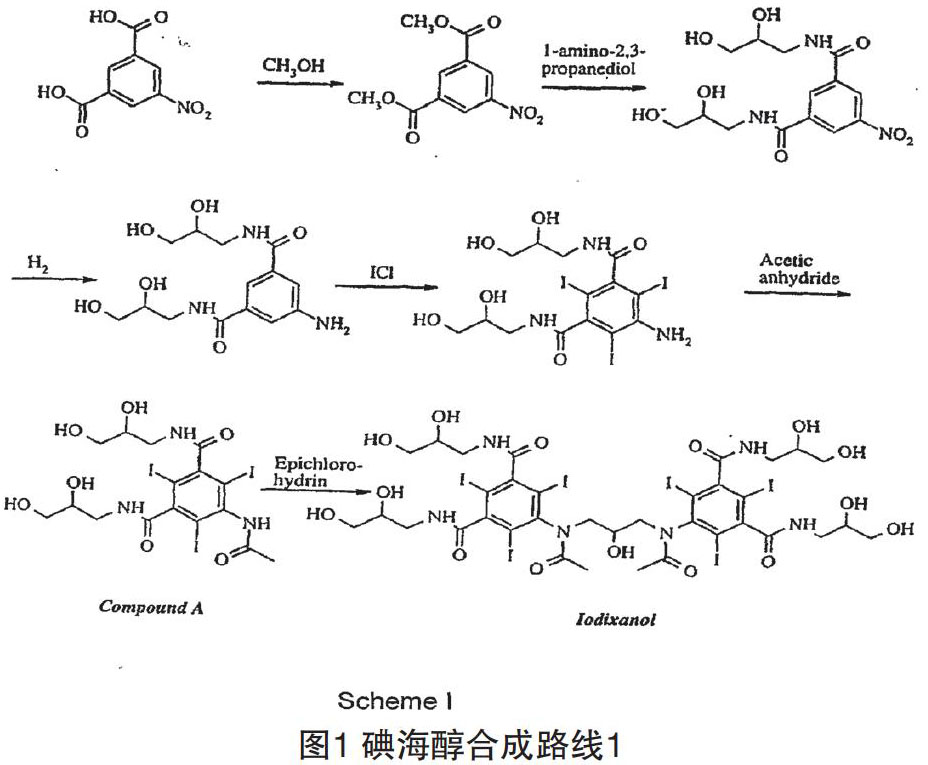
最早提出的技术路线是以5-硝基-1,3-二苯甲酸甲酯为原料,经酰胺化,还原,再酰化,最后N-烷基化而获得碘海醇粗品(如图1所示)。
这条路线的总收率较低,碘海醇原料药的纯化十分繁琐和昂贵[2]。
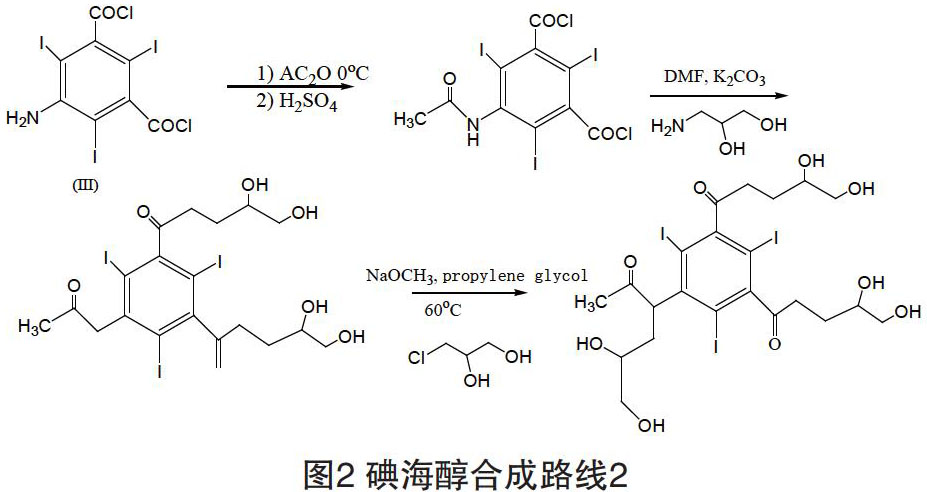
1.2 技术路线2
该条路线中,第一步反应的的收率是65%,第二步与1-氨基-2,3-丙二醇酰化收率是80%,与3-氯-1,2-丙二醇进行N-烷基化的收率是54%。
本专利技术对这条路线的合成工艺进行了改进,提高了单步反应的收率。按路线(如图2所示)合成碘海醇在技术和经济上更占优势[3]。
1.3 技术路线3
该技术路线(如图3所示)和前两条路线相比,收率有了较大提高,成本下降,操作安全性好[4]。
2 该项目采取的技术路线
2.1 合成碘海醇起始原料
技术路线3中所用的起始原料为5-氨基-2,4,6-三碘-1,3-二苯甲酰氯[5],但该原料价格昂贵,不利于降低碘海醇的制备成本,因此选用价格较低的5-硝基-1,3-二苯甲酸,经还原、碘化、酰氯化可以很方便得到起始原料5-氨基-2,4,6-三碘-1,3-二苯甲酰氯。
2.1.1 还原
该反应缺点是反应需在压力反应器密闭进行,时间较长(24h),操作上具有一定危险性。因此用以下方式进行改进与优化:
该反应在常压下进行,时间短(2h)左右,收率较高。
2.1.2 碘化
该反应的产率为80%左右。
2.1.3 合成二酰氯
该实验中使用过量的SOCl2,既作试剂也作溶剂,过量的SOCl2和催化剂可以蒸馏回收用于下批次反應。常用的催化剂是DMF[6],但DMF也会产生以下缺点:(1)需升温到90℃左右才能使反应完全,在此温度下,催化剂会部分分解,并和底物反应生成不溶的、组成不确定的副产物。(2)有文献报道DMF和SOCl2合用时,会形成有毒副产物(Levin, D. Org. Proc. Res. DeV. 1997, 1,182)。
因此,我们对催化剂进行优化,采用N-甲基吡咯烷酮或四甲基脲为催化剂,文献中报道这两种催化剂具有和DMF相似的催化活性、并可克服上述缺点。
2.2 碘海醇中间体Ⅰ、Ⅱ和原料药的制备
获得制备碘海醇的起始原料5-氨基-2,4,6-三碘基-1,
3-二苯甲酰氯后,采取以下路线来合成碘海醇中间体Ⅰ、Ⅱ和原料药。
2.2.1 中间体Ⅰ和5-酰胺二酰氯的合成
实施方案A:100g 5-氨基-2,4,6-三碘-1,3-二苯甲酰氯悬浮在500g冰醋酸中,缓慢加入60gSOCl2,反应3h,冷却后,所生成沉淀过滤回收,余液先用醋酸然后用水洗,母液中继续产生的沉淀和第一次所生成沉淀合并,可获得100g左右的产物,产率约93%(如图4所示)。
实施方案B:采用乙酰氯代替冰醋酸做酰化试剂,反应在双极性溶剂中进行,溶剂可选DMF,二甲基乙酰胺(DMA),二甲基亚砜(DMSO)或N-甲基吡咯烷酮。乙酰氯用量是二酰氯底物的1.5~3倍,反应时间24h以上,产物收集是将反应结束后的混合液滴入冰水中,沉淀出产物,过滤收集。反应产率和实施方案A相当。
2.2.2 中间体Ⅱ的合成
实施方案:370.4g5-乙酰-二酰氯(底物)溶于741.0gDMA,232.8g1-氨基-2,3-丙二醇溶于DMA,然后在0℃滴加入底物的DMA溶液中,两小时滴完(如图5所示)。反应在25℃反应7h进行完全。在真空度12mmHg,92℃蒸出DMA至干,冷却到50℃,残余物用950g甲醇和500g水溶解,混合物用7%NaOH溶液(质量浓度)调节pH值为10.5,并通过反复加入NaOH维持pH在10.5,使之形成5-乙酰氨基-N,N-双(2,3-二羟丙基)-2,4,6-1,3-苯二甲酰胺钠盐。将产物沉淀和1-氨基-2,3-丙二醇的回收后,反应液冷却到室温,加入36%HCl调节pH值到1.0,沉淀产物,过滤收集,干燥。母液浓缩近干,加入500g水,透过2000mLNa型Amberjet1200型阳离子交换树脂,1-氨基-2,3-丙二醇用7%氨水提取回收。
2.2.3碘海醇原料药的制备
实施方案:5-乙酰氨基-2,4,6-三碘-N,N-双(2,3-二羟丙基)-1,3-二苯甲酰胺,1,2-丙二醇为溶剂,加入甲醇钠甲醇溶液,50℃搅拌溶解,减压回收过量甲醇,冷却至室温,加入3-氯-1,2-丙二醇,室温搅拌48h。反应结束后,减压浓缩至干,冷却,向剩余物加入甲醇,过滤,除去不溶物,向滤液中加水,阳离子交换树脂,阴离子交换树脂,搅拌2h,滤去树脂,滤液减压浓缩至干,冷却,向剩余物加入正丁醇研磨,析出白色固体,得粗品,用正丁醇重结晶后的产率约82%(如图6所示)。
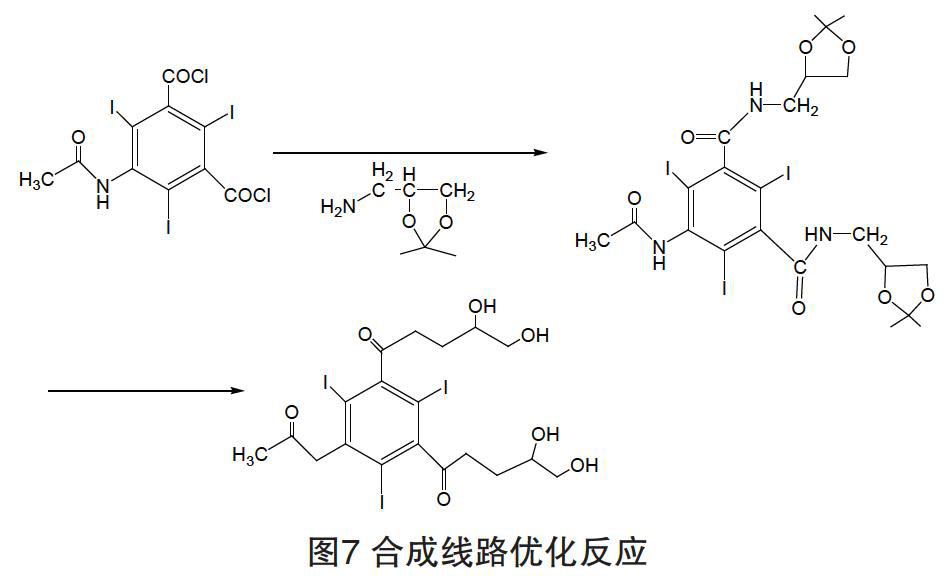
技术风险及障碍:在进行N-烷基化步骤时,会伴随O-烷基化,可能形成多种副产物,导致产物分离困难。
合成路线优化方案:在N-烷基化前在2,3位侧链烷基上引入氨基丙缩酮,在烷基化步骤后用阳离子交换树脂/甲醇-水体系脱去缩酮而获得碘海醇原料药(如图7所示)。
优点:预防O-烷基化副产物的产生,后处理中用阳离子释放H质子方式除去保护基,避免后处理中需采用重结晶来除去O-烷基化副产物。改进后反应时间缩短,操作简便安全,成本降低,能耗少。
参考文献
[1] 陈朝晖,董晓莉.离子色谱法测定食盐中的碘含量[J].现代科学仪器,2004(5):65.
[2] 陈昭平,罗来涛.酸处理对海泡石表面及其结构性质的影响[J].南昌大学学报(自然科学版),2000,24(1):68-72.
[3] 陈兆能,邱泽麟,余经洪.试验分析与设计[M].上海:上海交通大学出版社,1991.
[4] 黄宪.新编有机合成化学[M].北京:化工出版社,2003.
[5] 包丙男.近年来发展的非离子型水溶性有机碘造影剂[J].国外医药—合成药、生化药、制剂分册,1986,7(5):275-277.
[6] 吴恩惠.介绍经肾排泄水溶性碘造影剂[J].中华放射性杂志,1992,26(2):131-132.
- 企业并购及并购过程中投资银行的作用
- 粮食主产区融资渠道多样化研究
- 基于SWOT理论的众筹分析
- 众筹融资的风险分析与防范研究
- 辽宁省社区银行发展的外部配套条件及总体思路
- 经济政策不确定性和汇率波动
- 互联网金融对商业银行的影响研究及应对策略
- 我国商业银行经营效率的实证分析
- 汇率升降及其对我国经济的影响分析
- 探究基于风险管理的会计内部控制与完善
- 浅析我国注册会计师行业审计失败原因及防范措施
- 我国上市公司环境会计信息披露研究
- 社会责任会计研究方法综述
- 浅谈中小企业财务风险管理
- 浅谈如何加强财务预算管理
- 企业实施平衡计分卡的问题探析
- 基于XBRL技术下网络财务报告应用研究
- 企业应收账款管理策略探讨
- 监督全口径预算管理研究
- 企业社会责任在会计方面的研究
- 企业单位的应收账款管理与风险控制
- 基于财务视角的中国长寿企业可持续增长研究
- 民营企业财务管理存在的问题及对策分析
- 浅析企业同一控制下吸收合并特殊性税务处理
- 管理会计在商业银行应用的优化路径探析
- cultlike
- cults
- cultual
- cultural
- culturally
- culture
- cultured
- cultures
- culture shock
- culture-shocked
- cultureshocked
- culture shocks
- culture vulture
- culturing
- culvert
- culverts
- cum
- cumbersome
- cumbersomely
- cumbersomeness
- cumbersomenesses
- cumdividend
- cum dividend
- cume
- cumin
- 坻伏
- 坼
- 坼三台
- 坼中台
- 坼罅
- 坼裂
- 坽
- 坿
- 垂
- 垂下
- 垂下的帘子
- 垂下眼睑
- 垂世
- 垂世不朽
- 垂丝
- 垂丝海棠
- 垂两耳
- 垂临
- 垂于马腹两侧,用于遮挡尘土的东西
- 垂云
- 垂仁
- 垂佑
- 垂体
- 垂儽儽
- 垂光