色谱与质谱联用技术在蛋白质翻译后修饰研究中的进展及应用
林琳等
摘 要 蛋白质翻译后修饰是调控各种细胞信号通路和细胞命运最为关键的分子机制之一。在分子水平上,蛋白质翻译后修饰的“写入”、“擦除”和“识别”是实现其动态调控功能的核心过程,均属于生物分析化学的研究范畴。基于液相色谱与生物质谱联用的蛋白质组学技术经历了近十年的高速发展,已成为系统水平上表征各种蛋白质翻译后修饰的标准方法。本文综述了蛋白质翻译后修饰分析相关的关键技术和方法进展,主要包括:各种翻译后修饰蛋白质和多肽的富集方法、基于高效液相色谱的多维分离方法和各种生物质谱鉴定方法,并重点阐述了近年来的新的发展方向。基于相关技术的快速发展和大规模鉴定数据的产生,本文重点介绍了5种具有重要生物学功能的蛋白质翻译后修饰,包括:磷酸化、糖基化、泛素化、乙酰化和甲基化。生物分析化学领域的方法和技术创新必将进一步促进对蛋白质翻译后修饰的系统水平研究的发展。
关键词 蛋白质翻译后修饰; 蛋白质组学; 质谱; 综述
1 引 言
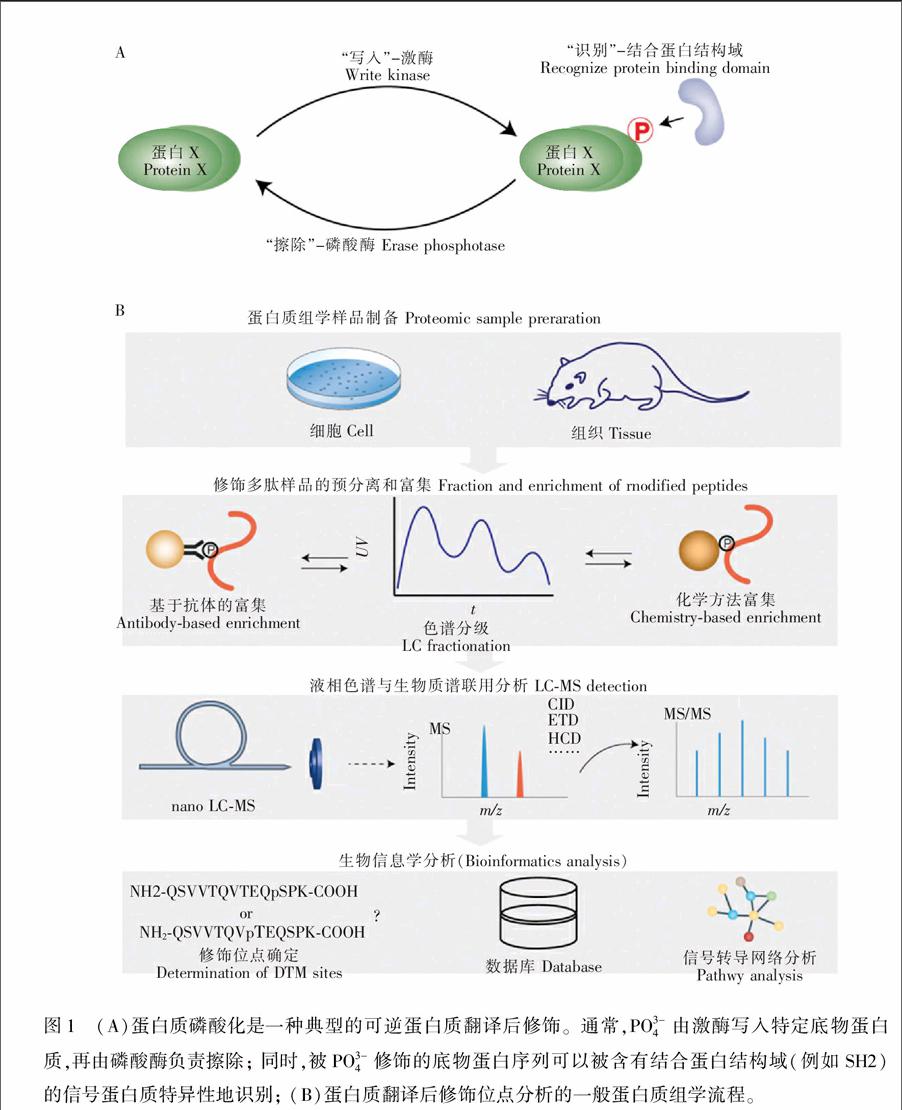
蛋白质翻译后修饰是蛋白质水平发生的一种重要的生物化学过程,可为蛋白质的特定氨基酸位点引入各种化学基团,例如磷酸根、糖基、甲基、乙酰基和泛素链等。在哺乳动物表达的所有蛋白质中, 超过50%的蛋白质可以在特定时间和亚细胞空间发生各种各样的翻译后修饰,并且调控着许多重要的生物学功能[1]。目前,已陆续发现近300种蛋白质翻译后修饰,其中很多重要的翻译后修饰都是以可逆方式被相关酶调控的[2]。这些可逆的蛋白质翻译后修饰是动态调控蛋白质功能的重要方式,同时也为生物体系提供了更高层次的复杂度。以具有重要生物学功能以及研究最为广泛的蛋白质磷酸化为例,使底物蛋白质发生磷酸化修饰的酶是激酶,而使磷酸化蛋白质发生去磷酸化的酶是磷酸酶。在人类基因组中,分别有518种激酶和约137种磷酸酶。激酶和磷酸酶常有多种底物蛋白质,而每种底物蛋白质往往有多个磷酸化位点。这些被磷酸化的氨基酸序列常会特异性地被蛋白结构域所识别(例如SH2、PTB、BRCT结构域等),从而形成基于磷酸化的蛋白质间相互作用[3,4]。如图1A所示,正是由于细胞内存在着数目庞大的可以“写入”、“擦除”和“识别”磷酸化位点的蛋白质,形成了一个极为复杂的由磷酸化介导的信号转导网络。从分子水平上,上述细胞内的生物化学过程主要涉及各种修饰基团的“写入”和“擦除”,以及蛋白质间基于特定修饰基团的“识别”。这类分子机理正是生物分析化学研究的核心生物学问题之一。
传统的蛋白质翻译后修饰研究主要依赖于基于特异性抗体的免疫检测技术或放射性标记技术。这些方法对研究由单一位点翻译后修饰介导的细胞信号转导过程起着不可替代的作用。然而,由于上述技术存在操作要求高、特异性抗体制备周期长等缺点,很难实现蛋白质翻译后修饰的大规模检测。近年来,基于液相色谱与生物质谱联用技术(LC-MS)的蛋白质组学策略发展迅速,为系统水平上的蛋白质翻译后修饰研究提供了强有力的研究工具[5,6]。应用于蛋白质翻译后修饰研究的研究策略与鉴定一般多肽样品的LC-MS流程是基本一致的。然而,蛋白质翻译后修饰研究常具有极大的技术挑战,这主要是因为:(1)被修饰蛋白质通常仅占总体蛋白质表达量的很小一部分; (2)翻译后修饰基团常具有化学稳定性差、在样品制备和LC-MS分析过程中容易丢失的缺点; (3)蛋白质翻译后修饰常是动态调节过程,这为样品制备和LC-MS分析提出了更高的要求。
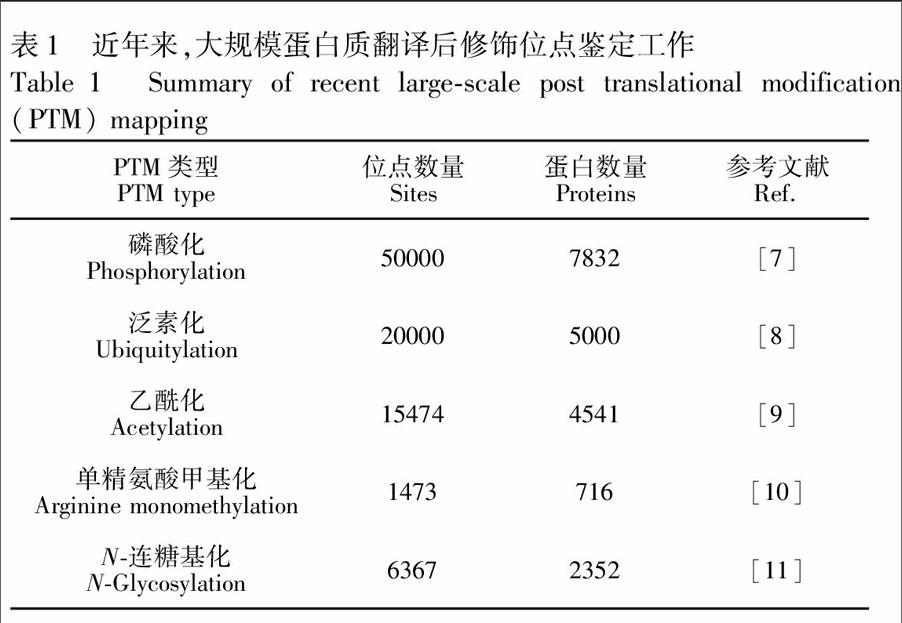
如图1B所示,蛋白质翻译后修饰位点分析的一般流程主要分为以下4个步骤:蛋白质组学样品制备、修饰多肽样品的预分离和富集、LC-MS分析和生物信息学分析。作为一种通用型分析技术策略,蛋白质组学技术可以实现对包括细胞、组织和体液在内的所有生物样品的全面LC-MS分析。近十年来,随着质谱硬件技术的快速发展,生物质谱的灵敏度和扫描速度都有了显著地提升; 与此同时,各种在线和离线色谱分离技术和针对多种重要翻译后修饰的选择性富集方法也层出不穷,为蛋白质翻译后修饰研究奠定了坚实的技术基础。目前,针对磷酸化等几种重要蛋白质翻译后修饰的蛋白质组学研究均可实现在几千到上万个位点的定性和定量表征(表1)。本文主要针对翻译后修饰蛋白及相应的酶解多肽的色谱富集技术、色谱分离技术及质谱检测技术进行
了综述。基于其生物学重要性和相关分析技术快速发展,本文重点综述蛋白质磷酸化、糖基化、泛素化、乙酰化和甲基化相关的研究进展,并对未来发展方向提出若干展望。
2 翻译后修饰蛋白及多肽的
富集方法
2.1 磷酸化修饰
磷酸化修饰是细胞信号转导和功能调控过程中最重要的翻译后修饰类型之一。由于磷酸化翻译后修饰的丰度极低,仅约占所在蛋白质总量的1%,因此,必须发展特异性的富集方法,以实现质谱的有效分析。目前,几种亲和富集方法发展较为完备,可以高效地从细胞酶解液中将磷酸化修饰的肽段富集出来,包括金属氧化物亲和色谱法(Metal oxide affinity chromatography,MOAC)、固定化金属离子亲和色谱法(Immobilized metal ion affinity chromatography,IMAC)和基于特异性抗体的富集方法。
2.1.1 金属氧化物亲和色谱法(MOAC) 金属氧化物亲和色谱是一类最常用和最为有效的磷酸化肽富集方法。基于金属氧化物对磷酸基团的高亲和力,可以实现对磷酸化多肽的高选择性富集(一般可以达90%以上)。多种金属氧化物已经被用于富集磷酸化肽,包括二氧化钛(TiO2)[12]、二氧化锆(ZrO2)[13],五氧化二铌(Nb2O5)[14]等,其中TiO2以其优异的富集效率和富集选择性被广泛使用。TiO2富集的一般流程包括以下步骤:首先将样品酸化(如0.1%(V/V)三氟乙酸),这个过程是为了将样品中的非磷酸化酸性肽段质子化,防止它们非特异性地吸附在TiO2颗粒上。然后将样品与TiO2颗粒充分接触,以实现对磷酸化肽的特异性吸附。经过洗涤步骤之后,磷酸化多肽在碱性条件下(如5%(V/V)氨水)从TiO2颗粒上洗脱下来。含有酸性氨基酸的多肽(如谷氨酸和天冬氨酸)常会竞争磷酸化肽的结合位点,从而降低富集方法的选择性。因此,上样缓冲液中常要加入2,5二羟苯甲酸[15]、邻苯二甲酸[16]或谷氨酸[17]等,有效抑制酸性多肽的吸附,从而提高富集选择性。
2.1.2 固定化金属离子亲和色谱法(IMAC) IMAC是另一种被广泛应用的磷酸化多肽富集方法。通常,正电性的金属离子可以被含有亚胺乙酸(IDA)或氨三乙酸(NTA)金属螯合基团的载体固定化,可以基于静电相互作用力, 可选择性地富集负电性的磷酸化多肽。各种各样的金属离子可以被固定化,并用于高效富集磷酸化多肽,例如Fe3+[18],Ti4+[19],Zr4+[20]等。IMAC的操作流程与MOAC类似:首先,多肽样品在酸性条件下上样,然后清洗非特异性吸附多肽,最后用高pH值的洗脱液或者磷酸盐缓冲液洗脱被富集的磷酸化多肽。
通常,IMAC富集法对多磷酸化肽的富集效果强于对单磷酸化肽的富集。因此,通过结合MOAC富集法,可以很好地实现对磷酸化多肽的全面富集。在Sequential elution from IMAC(SIMAC)方法中[21],基于Fe3+-IMAC的富集首先可以将多磷酸化肽很好地富集,然后再串联使用TiO2富集,以实现对大多数单磷酸化肽的全面富集。Zhou等[19,22]发展了一种新的高效磷酸化多肽富集方法Ti4+-IMAC。通过设计并合成多种含有固定化磷酸根的微球,可以实现对Ti4+等金属离子的高效螯合和磷酸化肽的高选择性富集。与其它4种常用磷酸化多肽富集方法(Fe3+-IMAC, Zr4+-IMAC, TiO2和ZrO2)相比,Ti4+-IMAC方法展示出更高的富集选择性和富集效率。该方法的高富集选择性主要是源于聚合物微球表面所含有的具有灵活性的间隔臂,以及Ti3+与PO34之间的高选择性作用力。
2.1.3 基于抗体的富集法 酪氨酸磷酸化发生率非常低,一般只约占人体全部磷酸化修饰的1%[23]。因此,发展可以区分丝氨酸和苏氨酸磷酸化肽的高选择性酪氨酸磷酸化肽富集方法就显得尤为必要。基于固定化抗体的富集方法是目前针对酪氨酸磷酸化肽最有效的富集方法。目前, 最有效的富集抗体包括4G10抗体和PY100抗体。以PY100抗体为例,Rikova等[24]从41种非小细胞肺癌细胞和150种非小细胞肺癌肿瘤中共富集并鉴定到2700多个蛋白和4551个酪氨酸磷酸化位点,是目前最大规模的酪氨酸磷酸化蛋白质组学分析。基于固定化抗体的富集方法的富集选择性一般较差,通过结合MOAC或IMAC富集方法可以大大提高富集选择性[7,25]。然而,引入额外的富集步骤也会大大增加微量样品的损失。在本研究组的前期工作中,通过系统优化抗体富集的各步骤,实现了一步抗体富集鉴定超过800个酪氨酸磷酸化位点[26]。
2.2 糖基化修饰
蛋白质糖基化修饰参与着很多重要的细胞功能,例如细胞粘附,受体膜蛋白激活,细胞免疫反应等。大多数位于细胞膜,内质网和细胞外的蛋白质均会发生糖基化修饰。在众多蛋白质糖基化修饰中,发生在天冬酰胺上的N-连糖基化修饰和发生在丝氨酸和苏氨酸上的O-连糖基化修饰是最常见和研究最广泛的糖基化修饰。应用于富集糖基化蛋白质或多肽的方法主要有固定化凝集素和酰肼化学两种。
2.2.1 固定化凝集素富集 基于凝集素对多糖的选择性吸附作用,将凝集素固定到合适的基质上可以实现对糖蛋白或者糖肽的特异性富集[27]。伴刀豆凝集素(Con A)、麦胚凝集素(WGA)、花生凝集素(PNA)和橙黄网孢盘菌凝集素(AAA)是针对N-连糖基化蛋白富集广泛使用的几种凝集素。由于不同凝集素识别不同的糖型,通过结合使用凝集素ConA、WGA、N-乙酰葡糖胺和凝集素RCA120, Zielinska等[11]发展了一种膜辅助样品处理方法用于糖基化蛋白的富集和糖基化位点鉴定,共从4种小鼠组织和血浆中鉴定5753个糖基化位点,是目前最大的N-连糖基化数据集。
2.2.2 基于酰肼化学的富集
另一种广泛使用的糖基化蛋白和糖肽富集方法是由Zhang等[28]发展的基于酰肼化学的N-连糖基化富集方法。其主要步骤是:首先将糖基化蛋白用高碘酸钠氧化; 被氧化糖链产生的醛基可以与含有酰肼基团的树脂发生共价键反应,从而实现对糖基化蛋白的高选择性富集; 所固定化的糖基化蛋白可以被糖苷水解酶酶解释放,可以用于后续的质谱分析。这种方法最大的优点是反应效率高和特异性好。最近,Zhang等[29]报道了一种基于氨氧基树脂的氧化糖基化蛋白富集方法。与传统的酰肼化学法相比,该方法具有更高的反应活性,从而大大降低了样品处理的周期。
2.3 泛素化修饰
泛素是含有76个氨基酸的多肽序列。一个或多个该多肽序列可以通过其C末端的两个甘氨酸残基选择性地修饰到多个底物蛋白质的赖氨酸位点上。蛋白质泛素化修饰是激活蛋白质降解机制的关键性信号,在DNA修复和细胞凋亡等重要生物学过程中都起着至关重要的作用。传统的泛素化蛋白质的富集主要通过在其泛素链末端引入亲和富集标签实现。以酵母细胞为模型和6×His tag标签对泛素化进行标记,Peng等[30]首次实现了对泛素化蛋白及其泛素化位点的蛋白质组学鉴定。然而,该方法的显著缺陷是缺乏对泛素化位点的选择性富集。基于胰蛋白酶酶解产生的泛素化多肽会残留两个甘氨酸残基的特征,目前最为有效的泛素化多肽富集方法是基于上述双甘氨酸残基的抗体富集法[31]。通过结合各种离线色谱分离策略,该抗体富集法已经被广泛应用于鉴定成千上万个泛素化位点[32]。
2.4 乙酰化修饰
蛋白质乙酰化修饰主要发生在蛋白质赖氨酸位点上,是组蛋白和转录因子等关键功能性蛋白质的重要调控机制。大规模乙酰化位点鉴定主要得益于特异性抗体和先进的质谱仪器的发展。芝加哥大学赵英明团队首先发展了一种针对乙酰化蛋白的抗体,并首次实现对蛋白质乙酰化位点的系统水平分析,共鉴定了388个乙酰化位点[33]。最近,Mann等[34]使用相同抗体并结合先进的轨道阱质谱仪(Orbitrap)实现了对乙酰化位点最大规模的鉴定,共从人类急性髓系白血病细胞器中鉴定到约3600个赖氨酸乙酰化位点。这充分说明了特异性抗体对低丰度修饰多肽富集的高效率和先进质谱技术对大规模蛋白质组学分析的重要性。
2.5 甲基化修饰
蛋白质甲基化修饰主要发生在蛋白质的赖氨酸和精氨酸位点上。与乙酰化修饰类似,蛋白质甲基化修饰也是DNA损伤修复、基因转录和信号转导的重要调控途径。然而,与其它类型翻译后修饰相比,蛋白质甲基化修饰的大规模蛋白质组学研究一直比较落后。这主要是因为甲基化修饰基团较小,很难发展针对其化学结构的有效富集方法。Boisvert等[35]首先使用4种特异性精氨酸甲基化抗体(ASYM24、ASYM25、SYM10和SYM11)对甲基化蛋白质进行了蛋白质组学分析。最近,Cell Signaling公司发展了多种针对不同类型甲基化的特异性抗体,实现了对超过1000多个精氨酸甲基化位点和约160个赖氨酸甲基化位点的大规模鉴定[10]。
正如引言中所述,大部分重要的蛋白质翻译后修饰都有相应的特异性蛋白质结合结构域[36]。例如,人蛋白质组中有120个可以特异性识别酪氨酸磷酸化的SH2蛋白质结合结构域。该结构域已经被广泛地应用于高选择性地检测和富集酪氨酸磷酸化蛋白质[37]。在人蛋白质组中也存在着5种甲基化蛋白质结合结构域: Tudor, Chromo, MBT, PWWP和Agenet[38]。最近, Moore等[39]筛选获得一种MBT结构域,可以实现对赖氨酸甲基化蛋白质的广谱性富集和大规模蛋白质组学分析。可以预期基于蛋白质结合结构域的亲和富集方法将在蛋白质翻译后修饰研究中起到越来越重要的作用。
2.6 逐步富集多种修饰蛋白
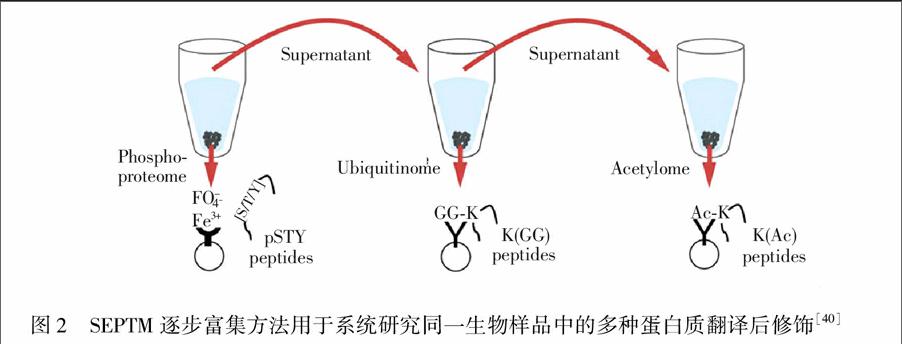
多种蛋白质翻译后修饰的协同调控是调控各种生物学过程的重要分子机制。实现对这种协同调控机制的系统水平表征,将为生物学研究提供更高维度的分子机理。最近Mertins等[40]发展了一种针对同一生物学样品中蛋白质磷酸化、乙酰化和泛素化修饰多肽的逐步富集方法(SEPTM),从而实现了对特定样品中多种蛋白质翻译后修饰的全面分析。该方法依次对同一生物样品使用IMAC亲和富集、泛素化特异性抗体富集和乙酰化特异性抗体富集,共检测到20000多个磷酸化修饰位点、15000多个泛素化修饰位点和3000多个乙酰化修饰位点(图2)。在这些被修饰的多肽中有0.3%的多肽存在多种修饰类型同时发生的现象。Swaney等[41]针对蛋白质磷酸化和泛素化修饰多肽进行了类似的串联富集和大规模质谱鉴定,最终发现在466个蛋白中同时存在2100个磷酸化修饰位点和2189个泛素化修饰位点。
图2 SEPTM逐步富集方法用于系统研究同一生物样品中的多种蛋白质翻译后修饰[40]
Fig.2 Serial enrichment of different post-translational modification (SEPTM) enables sequential enrichment of different PTMs from the same biological sample[40]
3 多维液相色谱分离
蛋白质组学样品均具有极高的复杂度,因此需要对其进行高效的分离以提高质谱鉴定效率。在蛋白质翻译后修饰研究中,在选择性富集修饰多肽前或者富集后常需要对酶解多肽进行有效的预分离。高效液相色谱是目前蛋白质组学策略中的标准化分离手段,这主要是因为其具有极高的酶解多肽分离能力,并且能很好地与质谱仪进行在线联用[42]。为了显著提高液相色谱的分离效率,降低多肽的复杂度,多维液相色谱是目前最为广泛使用的色谱分离模式[43]。基于在线低pH反相液相色谱与质谱仪之间的良好相容性,其它色谱分离模式与在线低pH反相液相色谱之间的的分离正交性是多维液相色谱分离中最重要的考虑因素[44]。以应用磷酸化蛋白质组学为例,介绍目前最为广泛使用的几种多维液相色谱分离模式。
3.1 离子交换色谱分离
离子交换色谱用于多肽分离主要是基于磷酸化多肽与固定相表面的强阳离子交换基团(SCX)或强阴离子交换基团(SAX)间的静电相互作用实现分离。另外,在高乙腈浓度流动相中,众多离子交换色谱填料可以展示出很好的亲水相互作用色谱(HILIC)性质,从而可以基于亲水相互作用分离酶解多肽[45]。在磷酸化蛋白质组学中,SCX是被最为广泛使用的一种色谱分离模式[18]。SCX分离磷酸化多肽的主要原理是:在酸性条件(如pH 2.7)下,磷酸化多肽的N端氨基和C端的赖氨酸或精氨酸残基都被质子化,磷酸基团的存在会降低多肽的价态。因此,磷酸化肽在SCX色谱填料上的保留较常规多肽更弱,从而实现了磷酸化多肽的有效富集和分离。通过与IMAC富集方法结合,Hottlin等[46]从9种小鼠组织中共鉴定到36000多个磷酸化位点,很好地展示了SCX与反相液相色谱的高正交性和其应用于磷酸化蛋白质组学研究的强大分离能力。与SCX相比,SAX对磷酸化肽具有更有效的保留,因此也被广泛地应用于磷酸化蛋白质组学研究中[47,48]。其主要优势是可以对多磷酸化肽、单磷酸化肽和非磷酸化肽实现更有效的分离。然而,离子交换色谱模式进行多肽分离一般都需要使用高浓度盐进行梯度洗脱,从而引入了额外的柱后除盐步骤,导致样品损失。
3.2 基于高pH反相液相色谱的分离
多肽在反相色谱中的分离是基于其与固定相上的C18基团之间的疏水相互作用实现的。Gilar等[50]首次将高pH反相色谱和低pH反向色谱结合使用,进行二维液相色谱分离,获得了优秀的正交性。该二维分离模式正交性的主要原因可能是肽段在不同的pH条件下电荷分布不同。中科院大连化物所邹汉法研究组对该方法在磷酸化多肽的分离方面进行了系统优化,并创造性地引入了等间隔先流出组分和后流出组分样品混合法(图3)[49]。该方法的优势是:(1)去除了额外的除盐步骤,从而大大降低了微量多肽样品的损失; (2)通过合并样品可以大大减少质谱分析时间,但同时又不降低二维色谱分离的正交性。正是由于上述显著的样品处理优势,该分离策略成为目前蛋白质组学领域中最为广泛使用的多维色谱分离模式[32,51]。基于上述介绍的多维色谱模式, Ficarro等[52]进一步发展了一种基于高pH反相色谱,SAX色谱和低pH反相色谱的三维色谱分离系统; 通过与质谱进行在线联用,最大化地实现了复杂磷酸化多肽样品的分离和高灵敏度质谱检测。作者从1×106~3×106个细胞中(约5~10 μg)共鉴定了约2500个非冗余的磷酸化多肽,是目前应用于蛋白质翻译后修饰研究最灵敏的LC-MS技术。
4 生物质谱鉴定
应用于蛋白质翻译后修饰的生物质谱技术主要需要完成以下任务:(1)鉴定发生修饰的蛋白质; (2)鉴定发生修饰的氨基酸位点及修饰基团的化学结构; (3)定量分析修饰蛋白占总体蛋白表达量的比例。存在于蛋白质上的修饰基团常具有化学稳定性差和丰度低的特点,因此对于质谱检测提出了更高的要求。近年来,多种温和的碎裂技术相继诞生,大大促进了修饰多肽的质谱检测效果。以研究最为广泛的磷酸化修饰为例,本文阐述应用于修饰多肽检测的重要质谱碎裂技术,主要包括:碰撞诱导解离(Collision-induced dissociation, CID)、高能碰撞解离技术(High-energy collisional dissociation,HCD)、基于电子诱导的电子捕获解离(Electron capture dissociation,ECD)和电子转移解离技术(Electron transfer dissociation,ETD)。
4.1 碰撞诱导解离(CID)
CID是串联质谱法中最常用的碰撞解离方法。它利用惰性气体分子撞击肽段母离子使其在酰胺键处断裂,产生具有丰富序列信息的b-和y-型离子。由于磷酸基团在CID过程中易发生中性丢失,影响了它对磷酸化多肽序列的有效鉴定。中性丢失的程度与很多因素有关,如电荷态、化学结构、质子化试剂、碎裂能量等。酪氨酸磷酸化易丢失HPO3(80 Da)的中性碎片,而丝氨酸和苏氨酸磷酸化更倾向于丢失H3PO4 (98 Da)的中性碎片[53]。这主要是由于酪氨酸磷酸化的CO键键能更大,不易碎裂。
为了解决磷酸化肽易发生中性丢失的问题,在离子阱质谱中常进一步采用第三级质谱MS3碎裂,以获得更多的碎片离子信息[54]。多级活化解离,又称虚拟的MS3,将发生中性丢失的产物离子和母离子同时活化,并记录所有的碎片离子[55]。MSA方法常用于低分辨的离子阱质谱中,而在高分辨、高质量精度类质谱(如Orbitrap)中的应用不多[54]。这主要是因为这类质谱可以高通量地获得母离子的精确质量数,因此常规的MS2碎裂已经可以满足检测需求,额外的MS3和多级活化解离反而会消耗本可以用于测序新肽段的质谱时间。
4.2 高能碰撞诱导解离(HCD)
HCD通过在高能碰撞室中提供比 CID更高的碰撞能量和更短的活化时间来解离离子[56,57]。与基于离子阱的CID相比,HCD所产生的中性丢失峰较少,主要产生序列特异性的y-型离子和某些b-型离子(图4)。在HCD中,b-型离子倾向于进一步解离成a-型离子,因此b-型离子的数目较y-型离子少。同时,由于HCD的能量更大,它常产生更小的碎片离子,如磷酸化酪氨酸特异性的Immonium离子。HCD碎裂克服了常规离子阱存在的1/3低质量范围限制,同时提供了比CID更高的分辨率,是Orbitrap质谱仪的标配解离模式,被广泛用于蛋白质翻译后修饰的研究。
4.3 基于电子诱导的解离(ECD/ETD)
与CID相比,基于电子诱导的解离方法,电子捕获解离(ECD)和电子转移解离(ETD),是一种温和的软解离方式,能够产生肽骨架碎裂信息并保留修饰基团。在ECD过程中,母离子被近热电子活化,酰胺键上的氧原子从邻近的氨基酸残基上转移一个质子,此过程放热使N-Cα键发生断裂产生c-型和z-型离子[58]。在ETD过程中,荧蒽自由基阴离子携带电子与肽段阳离子发生相互作用,此过程使多肽电荷数降低并诱导肽骨架的碎裂,产生一系列互补的c-型和z-型离子[59]。与CID相比,ETD更适合高电荷态和包含碱性残基肽段的测序。CID和ETD的互补使用有利于增加蛋白的鉴定数目和提高序列覆盖度,对大规模鉴定磷酸化肽十分有利[60]。另外,ECD和ETD方法是目前应用于糖基化多肽糖链结构解析最为有效的裂解方法[61]。基于上述的解离方法,Heck研究小组发展了一种集成化的解离技术,名为EThcD。顾名思义,该方法将适合磷酸化位点分析的ETD和HCD碎裂技术结合起来[62]。ETD不能断裂N-端连接脯氨酸残基的NCα键,这限制了它对脯氨酸含量高的磷酸化肽的修饰位点的确认。而EThcD方法很好地克服了这方面的缺陷,能够产生丰富的b/y和c/z型离子。
5 结论与展望
蛋白质翻译后修饰多达300多种,它们具有化学稳定性差、丰度低和动态变化等特点,在各种生物学过程中发挥着重要的调控作用。基于LC-MS的蛋白质组学技术已经实现了对成千上万种修饰位点的大规模定性和定量分析,这其中主要包括磷酸化、糖基化、泛素化、乙酰化和甲基化修饰。如本文所述,对这些重要翻译后修饰的大规模蛋白质组学分析的实现主要归功于众多方法学的巨大进步,主要包括高选择性和高效富集方法的发展、高容量和高正交性多维色谱技术的发展以及高灵敏度和高扫描速度生物质谱技术的发展。随着通用型多维色谱技术和生物质谱技术的不断完善,新的蛋白质翻译后修饰的大规模蛋白质组学研究更多局限于高选择性和高效富集方法的发展。如表1所示,在目前主要研究的蛋白质翻译后修饰中,甲基化修饰的大规模质谱鉴定仍处于较低的水平。这主要是因为各种甲基化修饰基团的分子尺寸较小,很难发展高特异性的富集方法。在传统抗体技术无法很好地解决这一难题的情况下,基于MBT甲基化蛋白质结合结构域的富集方法为甲基化蛋白质组研究提供了新颖的高效富集方法[39]。由于几乎所有的重要蛋白质翻译后修饰都有众多与之对应的蛋白质结合结构域,可以预期该类富集方法将会成为未来蛋白质翻译后修饰富集方法发展的重要方向。
此外,随着多种重要蛋白质翻译后修饰大规模蛋白质组学分析方法的逐步成熟,越来越多的研究者开始关注在相关生物体系中的应用研究,尤其是对多种翻译后修饰在同一蛋白质上的协同作用研究。目前的相关研究主要针对大规模质谱鉴定效果最好的翻译后修饰,即磷酸化、泛素化和乙酰化。可以预期,随着其它相关蛋白质翻译后修饰富集方法的不断发展和完善,针对特定生物学体系的多种翻译后修饰的协同作用研究将会得到进一步发展,并在相关生物学研究中起到关键作用。总之,色谱与质谱联用的技术已经为蛋白质翻译后修饰研究提供了革命性的系统水平研究工具,传统生物学领域的“一个基因,一个蛋白质,一种生物学功能”的理念已经被完全打破。基于色谱与质谱联用技术的蛋白质组学策略所产生的海量质谱数据, 已经推动蛋白质翻译后修饰及相关生物学领域进入了全新的系统生物学研究时代。
References
1 Scott J D, Pawson T. Science, 2009, 326: 1220-1224
2 Witze E S, Old W M, Resing K A, Ahn N G. Nat.Methods, 2007, 4: 798-806
3 Choudhary C, Mann M. Nat. Rev. Mol. Cell. Biol., 2010, 11: 427-439
4 Altelaar A F, Munoz J, Heck A J. Nat. Rev. Genet., 2013, 14: 35-48
5 Nilsson C L. Anal. Chem., 2012, 84: 735-746
6 Wang F, Song C, Cheng K, Jiang X, Ye M, Zou H. Anal. Chem., 2011, 83: 8078-8085
7 Sharma K, D′Souza R C, Tyanova S, Schaab C, Wisniewski J R, Cox J, Mann M. Cell Reports, 2014, 8: 1583-1594
8 Udeshi N D, Svinkina T, Mertins P, Kuhn E, Mani D R, Qiao J W, Carr S A. Mol. Cell. Proteomics, 2013, 12: 825-831
9 Lundby A, Lage K, Weinert B T, Bekker-Jensen D B, Secher A, Skovgaard T, Kelstrup C D, Dmytriyev A, Choudhary C, Lundby C, Olsen J V. Cell Reports, 2012, 2: 419-431
10 Guo A, Gu H, Zhou J, Mulhern D, Wang Y, Lee K A, Yang V, Aguiar M, Kornhauser J, Jia X, Ren J, Beausolei S A, Silva J C, Vemulapalli V, Bedford M T, Comb M J. Mol. Cell. Proteomics, 2014, 13: 372-387
11 Zielinska D F, Gnad F, Wisniewski J R, Mann M. Cell, 2010, 141: 897-907
12 Olsen J V, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Cell, 2006, 127: 635-648
13 Sugiyama N, Masuda T, Shinoda K, Nakamura A, Tomita M, Ishihama Y. Mol. Cell. Proteomics, 2007, 6: 1103-1109
14 Ficarro S B, Parikh J R, Blank N C, Marto J A. Anal.Chem., 2008, 80: 4606-4613
15 Larsen M R, Thingholm T E, Jensen O N, Roepstorff P, Jrgensen T J D. Mol. Cell. Proteomics, 2005, 4: 873-886
16 Bodenmiller B, Mueller L N, Mueller M, Domon B, Aebersold R. Nat. Methods, 2007, 4: 231-237
17 Wu J, Shakey Q, Liu W, Schuller A, Follettie M T. J. Proteome Res., 2007, 6: 4684-4689
18 Villen J, Gygi S P. Nat. Protocols, 2008, 3: 1630-1638
19 Zhou H, Ye M, Dong J, Corradini E, Cristobal A, Heck A J, Zou H F, Mohammed S. Nat. Protocols, 2013, 8: 461-480
20 Feng S, Ye M L, Zhou H J, Jiang X G, Jiang X N, Zou H F,Gong B. Mol. Cell. Proteomics, 2007, 6: 1656-1665
21 Thingholm T E, Jensen O N, Robinson P J, Larsen M R. Mol. Cell. Proteomics, 2008, 7: 661-671
22 Zhou H, Ye M, Dong J, Han G, Jiang X, Wu R, Zou H F. J.Proteome Res., 2008, 7: 3957-3967
23 Hunter T, Sefton B M. Proc. Natl. Acad. Sci. U. S. A., 1980, 77: 1311-1315
24 Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H. Cell, 2007, 131: 1190-1203
25 Jorgensen C, Sherman A, Chen G I, Pasculescu A, Poliakov A, Hsiung M, Larsen, B, Wilkinson D G, Linding R, Pawson T. Science, 2009, 326: 1502-1509
26 Tian R, Wang H, Gish G D, Petsalaki E, Pasculescu A, Shi Y, Mollenauerd M, Bagshawa R D, Yoseff N, Huntere T, Gingrasa A C,Weissd A,Pawsona T. Proc. Natl. Acad. Sci. U. S. A., 2015, 112: E1594-E1603
27 Kaji H, Saito H, Yamauchi Y, Shinkawa T, Taoka M, Hirabayashi J, Kasai K, Takahashi N, Isobel T. Nat. Biotechnol., 2003, 21: 667-672
28 Zhang H, Li X J, Martin D B, Aebersold R. Nat. Biotechnol., 2003, 21: 660-666
29 Zhang Y, Yu M, Zhang C, Ma W, Zhang Y, Wang C, Lu H. Anal.Chem., 2014, 86(15): 7920-7924
30 Peng J, Schwartz D, Elias J E, Thoreen C C, Cheng D, Marsischky G, Roelofs J, Finley D, Gygi S P. Nat. Biotech., 2003, 21: 921-926
31 Kim W, Bennett E J, Huttlin E L, Guo A, Li J, Possemato A, Sowa M, Rad R, Rush J, Comb M J, Harper J W, Gygi S P. Mol. Cell, 2011, 44: 325-340
32 Udeshi N D, Mertins P, Svinkina T, Carr SA. Nat.Protocols, 2013, 8: 1950-1960
33 Kim S C, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao Hao, Lin X, Grishin N V,White M, Yang X J, Zhao Y. Mol.Cell, 2006, 23: 607-618
34 Choudhary C, Kumar C, Gnad F, Nielsen M L, Rehman M, Walther T C,Olsen J V, Mann M. Science, 2009, 325: 834-840
35 Boisvert F M, Cté J, Boulanger M C, Richard S. Mol. Cell. Proteomics, 2003, 2: 1319-1330
36 Seet B T, Dikic I, Zhou M M, Pawson T. Nature Reviews Mol. Cell.Biol., 2006, 7: 473-483
37 Machida K, Thompson C M, Dierck K, Jablonowski K, Karkkainen S, Liu B, Zhang H, Nash P D, Newman D K, Nollau P, Pawson T, Renkema G H, Saksela K, Schiller M R, Shin D G, Mayer B J. Mol.Cell, 2007, 26: 899-915
38 Chen C, Nott T J, Jin J, Pawson T. Nat. Rev. Mol.Cell.Biol., 2011, 12: 629-642
39 Moore K E, Carlson S M, Camp N D, Cheung P, James R G, Chua K F,Wolf-Yadlin A, Gozani O. Mol. Cell, 2013, 50: 444-456
40 Mertins P, Qiao J W, Patel J, Udeshi N D, Clauser K R, Mani D R, Burgess M W, Gillette M A, JaffeJ D, Carr S A. Nat. Methods, 2013, 10: 634-637
41 Swaney D L, Beltrao P, Starita L, Guo A, Rush J, Fields S, Krogan N J, Villén J. Nat. Methods, 2013, 10: 676-682
42 Link A J, Eng J, Schieltz D M, Carmack E, Mize G J, Morris D R, Garvik B M, Yates J R. Nat. Biotechnol., 1999, 17: 676-682
43 Motoyama A, Yates J R, 3rd. Anal. Chem., 2008, 80: 7187-7193
44 Gilar M, Olivova P, Daly A E, Gebler J C. Anal. Chem., 2005, 77: 6426-6434
45 Boersema P J, Mohammed S, Heck A J. Anal. Bioanal. Chem., 2008, 391: 151-159
46 Huttlin E L, Jedrychowski M P, Elias J E, Goswami T, Rad R, Beausoleil S A, Villén J, Haas W, Sowa M E, Gygi S P. Cell, 2010, 143: 1174-1189
47 Han G, Ye M, Zhou H, Jiang X, Feng S, Jiang X, Tian R, Wan D, Zou H, Gu J. Proteomics, 2008, 8: 1346-1361
48 Wisniewski J R, Nagaraj N, Zougman A, Gnad F, Mann M. J. Proteome Res., 2010, 9: 3280-3289
49 Song C X, Ye M L, Han G H, Jiang X N, Wang F J, Yu Z Y, Zou H. Anal. Chem., 2010, 82(1): 53-56
50 Gilar M, Olivova P, Daly A E, Gebler J C. J. Sep. Sci., 2005, 28: 1694-1703
51 Kim M S, Pinto S M, Getnet D, Nirujogi R S, Manda S S, Chaerkady R. Nature, 2014, 509: 575-581
52 Ficarro S B, Zhang Y, Carrasco-Alfonso M J, Garg B, Adelmant G, Webber J T. Mol. Cell. Proteomics, 2011, 10: O111 011064
53 Boersema P J, Mohammed S, Heck A J R. J. Mass Spec., 2009, 44: 861-878
54 Villen J, Beausoleil S A, Gygi S P. Proteomics, 2008, 8: 4444-4452
55 Schroeder M J, Shabanowitz J, Schwartz J C, Hunt D F, Coon J J. Anal. Chem., 2004, 76(13): 3590-3598
56 Olsen J V, Macek B, Lange O, Makarov A, Horning S, Mann M. Nat. Methods, 2007, 4: 709-712
57 Jedrychowski M P, Huttlin E L, Haas W, Sowa M E, Rad R, Gygi S P. Mol. Cell. Proteomics, 2011, 10: M111 009910
58 Syrstad E A, Turecek F. J. Am. Soc. Mass Spec., 2005, 16: 208-224
59 Chi A, Huttenhower C, Geer L Y, Coon J J, Syka J E P, Bai D L. Proc. Natl. Acad. Sci. U. S. A., 2007, 104: 2193-2198
60 Phanstiel D H, Brumbaugh J, Wenger C D, Tian S, Probasco M D, Bailey D J. Nat. Methods, 2011, 8: 821-827
61 Zhu Z, Su X, Clark D F, Go E P, Desaire H. Anal. Chem., 2013, 85: 8403-8411
62 Frese C K, Zhou H, Taus T, Altelaar A F M, Mechter K, Heck A J R. J.Proteome Res., 2013, 12: 1520-1525
摘 要 蛋白质翻译后修饰是调控各种细胞信号通路和细胞命运最为关键的分子机制之一。在分子水平上,蛋白质翻译后修饰的“写入”、“擦除”和“识别”是实现其动态调控功能的核心过程,均属于生物分析化学的研究范畴。基于液相色谱与生物质谱联用的蛋白质组学技术经历了近十年的高速发展,已成为系统水平上表征各种蛋白质翻译后修饰的标准方法。本文综述了蛋白质翻译后修饰分析相关的关键技术和方法进展,主要包括:各种翻译后修饰蛋白质和多肽的富集方法、基于高效液相色谱的多维分离方法和各种生物质谱鉴定方法,并重点阐述了近年来的新的发展方向。基于相关技术的快速发展和大规模鉴定数据的产生,本文重点介绍了5种具有重要生物学功能的蛋白质翻译后修饰,包括:磷酸化、糖基化、泛素化、乙酰化和甲基化。生物分析化学领域的方法和技术创新必将进一步促进对蛋白质翻译后修饰的系统水平研究的发展。
关键词 蛋白质翻译后修饰; 蛋白质组学; 质谱; 综述
1 引 言
蛋白质翻译后修饰是蛋白质水平发生的一种重要的生物化学过程,可为蛋白质的特定氨基酸位点引入各种化学基团,例如磷酸根、糖基、甲基、乙酰基和泛素链等。在哺乳动物表达的所有蛋白质中, 超过50%的蛋白质可以在特定时间和亚细胞空间发生各种各样的翻译后修饰,并且调控着许多重要的生物学功能[1]。目前,已陆续发现近300种蛋白质翻译后修饰,其中很多重要的翻译后修饰都是以可逆方式被相关酶调控的[2]。这些可逆的蛋白质翻译后修饰是动态调控蛋白质功能的重要方式,同时也为生物体系提供了更高层次的复杂度。以具有重要生物学功能以及研究最为广泛的蛋白质磷酸化为例,使底物蛋白质发生磷酸化修饰的酶是激酶,而使磷酸化蛋白质发生去磷酸化的酶是磷酸酶。在人类基因组中,分别有518种激酶和约137种磷酸酶。激酶和磷酸酶常有多种底物蛋白质,而每种底物蛋白质往往有多个磷酸化位点。这些被磷酸化的氨基酸序列常会特异性地被蛋白结构域所识别(例如SH2、PTB、BRCT结构域等),从而形成基于磷酸化的蛋白质间相互作用[3,4]。如图1A所示,正是由于细胞内存在着数目庞大的可以“写入”、“擦除”和“识别”磷酸化位点的蛋白质,形成了一个极为复杂的由磷酸化介导的信号转导网络。从分子水平上,上述细胞内的生物化学过程主要涉及各种修饰基团的“写入”和“擦除”,以及蛋白质间基于特定修饰基团的“识别”。这类分子机理正是生物分析化学研究的核心生物学问题之一。
传统的蛋白质翻译后修饰研究主要依赖于基于特异性抗体的免疫检测技术或放射性标记技术。这些方法对研究由单一位点翻译后修饰介导的细胞信号转导过程起着不可替代的作用。然而,由于上述技术存在操作要求高、特异性抗体制备周期长等缺点,很难实现蛋白质翻译后修饰的大规模检测。近年来,基于液相色谱与生物质谱联用技术(LC-MS)的蛋白质组学策略发展迅速,为系统水平上的蛋白质翻译后修饰研究提供了强有力的研究工具[5,6]。应用于蛋白质翻译后修饰研究的研究策略与鉴定一般多肽样品的LC-MS流程是基本一致的。然而,蛋白质翻译后修饰研究常具有极大的技术挑战,这主要是因为:(1)被修饰蛋白质通常仅占总体蛋白质表达量的很小一部分; (2)翻译后修饰基团常具有化学稳定性差、在样品制备和LC-MS分析过程中容易丢失的缺点; (3)蛋白质翻译后修饰常是动态调节过程,这为样品制备和LC-MS分析提出了更高的要求。
如图1B所示,蛋白质翻译后修饰位点分析的一般流程主要分为以下4个步骤:蛋白质组学样品制备、修饰多肽样品的预分离和富集、LC-MS分析和生物信息学分析。作为一种通用型分析技术策略,蛋白质组学技术可以实现对包括细胞、组织和体液在内的所有生物样品的全面LC-MS分析。近十年来,随着质谱硬件技术的快速发展,生物质谱的灵敏度和扫描速度都有了显著地提升; 与此同时,各种在线和离线色谱分离技术和针对多种重要翻译后修饰的选择性富集方法也层出不穷,为蛋白质翻译后修饰研究奠定了坚实的技术基础。目前,针对磷酸化等几种重要蛋白质翻译后修饰的蛋白质组学研究均可实现在几千到上万个位点的定性和定量表征(表1)。本文主要针对翻译后修饰蛋白及相应的酶解多肽的色谱富集技术、色谱分离技术及质谱检测技术进行
了综述。基于其生物学重要性和相关分析技术快速发展,本文重点综述蛋白质磷酸化、糖基化、泛素化、乙酰化和甲基化相关的研究进展,并对未来发展方向提出若干展望。
2 翻译后修饰蛋白及多肽的
富集方法
2.1 磷酸化修饰
磷酸化修饰是细胞信号转导和功能调控过程中最重要的翻译后修饰类型之一。由于磷酸化翻译后修饰的丰度极低,仅约占所在蛋白质总量的1%,因此,必须发展特异性的富集方法,以实现质谱的有效分析。目前,几种亲和富集方法发展较为完备,可以高效地从细胞酶解液中将磷酸化修饰的肽段富集出来,包括金属氧化物亲和色谱法(Metal oxide affinity chromatography,MOAC)、固定化金属离子亲和色谱法(Immobilized metal ion affinity chromatography,IMAC)和基于特异性抗体的富集方法。
2.1.1 金属氧化物亲和色谱法(MOAC) 金属氧化物亲和色谱是一类最常用和最为有效的磷酸化肽富集方法。基于金属氧化物对磷酸基团的高亲和力,可以实现对磷酸化多肽的高选择性富集(一般可以达90%以上)。多种金属氧化物已经被用于富集磷酸化肽,包括二氧化钛(TiO2)[12]、二氧化锆(ZrO2)[13],五氧化二铌(Nb2O5)[14]等,其中TiO2以其优异的富集效率和富集选择性被广泛使用。TiO2富集的一般流程包括以下步骤:首先将样品酸化(如0.1%(V/V)三氟乙酸),这个过程是为了将样品中的非磷酸化酸性肽段质子化,防止它们非特异性地吸附在TiO2颗粒上。然后将样品与TiO2颗粒充分接触,以实现对磷酸化肽的特异性吸附。经过洗涤步骤之后,磷酸化多肽在碱性条件下(如5%(V/V)氨水)从TiO2颗粒上洗脱下来。含有酸性氨基酸的多肽(如谷氨酸和天冬氨酸)常会竞争磷酸化肽的结合位点,从而降低富集方法的选择性。因此,上样缓冲液中常要加入2,5二羟苯甲酸[15]、邻苯二甲酸[16]或谷氨酸[17]等,有效抑制酸性多肽的吸附,从而提高富集选择性。
2.1.2 固定化金属离子亲和色谱法(IMAC) IMAC是另一种被广泛应用的磷酸化多肽富集方法。通常,正电性的金属离子可以被含有亚胺乙酸(IDA)或氨三乙酸(NTA)金属螯合基团的载体固定化,可以基于静电相互作用力, 可选择性地富集负电性的磷酸化多肽。各种各样的金属离子可以被固定化,并用于高效富集磷酸化多肽,例如Fe3+[18],Ti4+[19],Zr4+[20]等。IMAC的操作流程与MOAC类似:首先,多肽样品在酸性条件下上样,然后清洗非特异性吸附多肽,最后用高pH值的洗脱液或者磷酸盐缓冲液洗脱被富集的磷酸化多肽。
通常,IMAC富集法对多磷酸化肽的富集效果强于对单磷酸化肽的富集。因此,通过结合MOAC富集法,可以很好地实现对磷酸化多肽的全面富集。在Sequential elution from IMAC(SIMAC)方法中[21],基于Fe3+-IMAC的富集首先可以将多磷酸化肽很好地富集,然后再串联使用TiO2富集,以实现对大多数单磷酸化肽的全面富集。Zhou等[19,22]发展了一种新的高效磷酸化多肽富集方法Ti4+-IMAC。通过设计并合成多种含有固定化磷酸根的微球,可以实现对Ti4+等金属离子的高效螯合和磷酸化肽的高选择性富集。与其它4种常用磷酸化多肽富集方法(Fe3+-IMAC, Zr4+-IMAC, TiO2和ZrO2)相比,Ti4+-IMAC方法展示出更高的富集选择性和富集效率。该方法的高富集选择性主要是源于聚合物微球表面所含有的具有灵活性的间隔臂,以及Ti3+与PO34之间的高选择性作用力。
2.1.3 基于抗体的富集法 酪氨酸磷酸化发生率非常低,一般只约占人体全部磷酸化修饰的1%[23]。因此,发展可以区分丝氨酸和苏氨酸磷酸化肽的高选择性酪氨酸磷酸化肽富集方法就显得尤为必要。基于固定化抗体的富集方法是目前针对酪氨酸磷酸化肽最有效的富集方法。目前, 最有效的富集抗体包括4G10抗体和PY100抗体。以PY100抗体为例,Rikova等[24]从41种非小细胞肺癌细胞和150种非小细胞肺癌肿瘤中共富集并鉴定到2700多个蛋白和4551个酪氨酸磷酸化位点,是目前最大规模的酪氨酸磷酸化蛋白质组学分析。基于固定化抗体的富集方法的富集选择性一般较差,通过结合MOAC或IMAC富集方法可以大大提高富集选择性[7,25]。然而,引入额外的富集步骤也会大大增加微量样品的损失。在本研究组的前期工作中,通过系统优化抗体富集的各步骤,实现了一步抗体富集鉴定超过800个酪氨酸磷酸化位点[26]。
2.2 糖基化修饰
蛋白质糖基化修饰参与着很多重要的细胞功能,例如细胞粘附,受体膜蛋白激活,细胞免疫反应等。大多数位于细胞膜,内质网和细胞外的蛋白质均会发生糖基化修饰。在众多蛋白质糖基化修饰中,发生在天冬酰胺上的N-连糖基化修饰和发生在丝氨酸和苏氨酸上的O-连糖基化修饰是最常见和研究最广泛的糖基化修饰。应用于富集糖基化蛋白质或多肽的方法主要有固定化凝集素和酰肼化学两种。
2.2.1 固定化凝集素富集 基于凝集素对多糖的选择性吸附作用,将凝集素固定到合适的基质上可以实现对糖蛋白或者糖肽的特异性富集[27]。伴刀豆凝集素(Con A)、麦胚凝集素(WGA)、花生凝集素(PNA)和橙黄网孢盘菌凝集素(AAA)是针对N-连糖基化蛋白富集广泛使用的几种凝集素。由于不同凝集素识别不同的糖型,通过结合使用凝集素ConA、WGA、N-乙酰葡糖胺和凝集素RCA120, Zielinska等[11]发展了一种膜辅助样品处理方法用于糖基化蛋白的富集和糖基化位点鉴定,共从4种小鼠组织和血浆中鉴定5753个糖基化位点,是目前最大的N-连糖基化数据集。
2.2.2 基于酰肼化学的富集
另一种广泛使用的糖基化蛋白和糖肽富集方法是由Zhang等[28]发展的基于酰肼化学的N-连糖基化富集方法。其主要步骤是:首先将糖基化蛋白用高碘酸钠氧化; 被氧化糖链产生的醛基可以与含有酰肼基团的树脂发生共价键反应,从而实现对糖基化蛋白的高选择性富集; 所固定化的糖基化蛋白可以被糖苷水解酶酶解释放,可以用于后续的质谱分析。这种方法最大的优点是反应效率高和特异性好。最近,Zhang等[29]报道了一种基于氨氧基树脂的氧化糖基化蛋白富集方法。与传统的酰肼化学法相比,该方法具有更高的反应活性,从而大大降低了样品处理的周期。
2.3 泛素化修饰
泛素是含有76个氨基酸的多肽序列。一个或多个该多肽序列可以通过其C末端的两个甘氨酸残基选择性地修饰到多个底物蛋白质的赖氨酸位点上。蛋白质泛素化修饰是激活蛋白质降解机制的关键性信号,在DNA修复和细胞凋亡等重要生物学过程中都起着至关重要的作用。传统的泛素化蛋白质的富集主要通过在其泛素链末端引入亲和富集标签实现。以酵母细胞为模型和6×His tag标签对泛素化进行标记,Peng等[30]首次实现了对泛素化蛋白及其泛素化位点的蛋白质组学鉴定。然而,该方法的显著缺陷是缺乏对泛素化位点的选择性富集。基于胰蛋白酶酶解产生的泛素化多肽会残留两个甘氨酸残基的特征,目前最为有效的泛素化多肽富集方法是基于上述双甘氨酸残基的抗体富集法[31]。通过结合各种离线色谱分离策略,该抗体富集法已经被广泛应用于鉴定成千上万个泛素化位点[32]。
2.4 乙酰化修饰
蛋白质乙酰化修饰主要发生在蛋白质赖氨酸位点上,是组蛋白和转录因子等关键功能性蛋白质的重要调控机制。大规模乙酰化位点鉴定主要得益于特异性抗体和先进的质谱仪器的发展。芝加哥大学赵英明团队首先发展了一种针对乙酰化蛋白的抗体,并首次实现对蛋白质乙酰化位点的系统水平分析,共鉴定了388个乙酰化位点[33]。最近,Mann等[34]使用相同抗体并结合先进的轨道阱质谱仪(Orbitrap)实现了对乙酰化位点最大规模的鉴定,共从人类急性髓系白血病细胞器中鉴定到约3600个赖氨酸乙酰化位点。这充分说明了特异性抗体对低丰度修饰多肽富集的高效率和先进质谱技术对大规模蛋白质组学分析的重要性。
2.5 甲基化修饰
蛋白质甲基化修饰主要发生在蛋白质的赖氨酸和精氨酸位点上。与乙酰化修饰类似,蛋白质甲基化修饰也是DNA损伤修复、基因转录和信号转导的重要调控途径。然而,与其它类型翻译后修饰相比,蛋白质甲基化修饰的大规模蛋白质组学研究一直比较落后。这主要是因为甲基化修饰基团较小,很难发展针对其化学结构的有效富集方法。Boisvert等[35]首先使用4种特异性精氨酸甲基化抗体(ASYM24、ASYM25、SYM10和SYM11)对甲基化蛋白质进行了蛋白质组学分析。最近,Cell Signaling公司发展了多种针对不同类型甲基化的特异性抗体,实现了对超过1000多个精氨酸甲基化位点和约160个赖氨酸甲基化位点的大规模鉴定[10]。
正如引言中所述,大部分重要的蛋白质翻译后修饰都有相应的特异性蛋白质结合结构域[36]。例如,人蛋白质组中有120个可以特异性识别酪氨酸磷酸化的SH2蛋白质结合结构域。该结构域已经被广泛地应用于高选择性地检测和富集酪氨酸磷酸化蛋白质[37]。在人蛋白质组中也存在着5种甲基化蛋白质结合结构域: Tudor, Chromo, MBT, PWWP和Agenet[38]。最近, Moore等[39]筛选获得一种MBT结构域,可以实现对赖氨酸甲基化蛋白质的广谱性富集和大规模蛋白质组学分析。可以预期基于蛋白质结合结构域的亲和富集方法将在蛋白质翻译后修饰研究中起到越来越重要的作用。
2.6 逐步富集多种修饰蛋白
多种蛋白质翻译后修饰的协同调控是调控各种生物学过程的重要分子机制。实现对这种协同调控机制的系统水平表征,将为生物学研究提供更高维度的分子机理。最近Mertins等[40]发展了一种针对同一生物学样品中蛋白质磷酸化、乙酰化和泛素化修饰多肽的逐步富集方法(SEPTM),从而实现了对特定样品中多种蛋白质翻译后修饰的全面分析。该方法依次对同一生物样品使用IMAC亲和富集、泛素化特异性抗体富集和乙酰化特异性抗体富集,共检测到20000多个磷酸化修饰位点、15000多个泛素化修饰位点和3000多个乙酰化修饰位点(图2)。在这些被修饰的多肽中有0.3%的多肽存在多种修饰类型同时发生的现象。Swaney等[41]针对蛋白质磷酸化和泛素化修饰多肽进行了类似的串联富集和大规模质谱鉴定,最终发现在466个蛋白中同时存在2100个磷酸化修饰位点和2189个泛素化修饰位点。
图2 SEPTM逐步富集方法用于系统研究同一生物样品中的多种蛋白质翻译后修饰[40]
Fig.2 Serial enrichment of different post-translational modification (SEPTM) enables sequential enrichment of different PTMs from the same biological sample[40]
3 多维液相色谱分离
蛋白质组学样品均具有极高的复杂度,因此需要对其进行高效的分离以提高质谱鉴定效率。在蛋白质翻译后修饰研究中,在选择性富集修饰多肽前或者富集后常需要对酶解多肽进行有效的预分离。高效液相色谱是目前蛋白质组学策略中的标准化分离手段,这主要是因为其具有极高的酶解多肽分离能力,并且能很好地与质谱仪进行在线联用[42]。为了显著提高液相色谱的分离效率,降低多肽的复杂度,多维液相色谱是目前最为广泛使用的色谱分离模式[43]。基于在线低pH反相液相色谱与质谱仪之间的良好相容性,其它色谱分离模式与在线低pH反相液相色谱之间的的分离正交性是多维液相色谱分离中最重要的考虑因素[44]。以应用磷酸化蛋白质组学为例,介绍目前最为广泛使用的几种多维液相色谱分离模式。
3.1 离子交换色谱分离
离子交换色谱用于多肽分离主要是基于磷酸化多肽与固定相表面的强阳离子交换基团(SCX)或强阴离子交换基团(SAX)间的静电相互作用实现分离。另外,在高乙腈浓度流动相中,众多离子交换色谱填料可以展示出很好的亲水相互作用色谱(HILIC)性质,从而可以基于亲水相互作用分离酶解多肽[45]。在磷酸化蛋白质组学中,SCX是被最为广泛使用的一种色谱分离模式[18]。SCX分离磷酸化多肽的主要原理是:在酸性条件(如pH 2.7)下,磷酸化多肽的N端氨基和C端的赖氨酸或精氨酸残基都被质子化,磷酸基团的存在会降低多肽的价态。因此,磷酸化肽在SCX色谱填料上的保留较常规多肽更弱,从而实现了磷酸化多肽的有效富集和分离。通过与IMAC富集方法结合,Hottlin等[46]从9种小鼠组织中共鉴定到36000多个磷酸化位点,很好地展示了SCX与反相液相色谱的高正交性和其应用于磷酸化蛋白质组学研究的强大分离能力。与SCX相比,SAX对磷酸化肽具有更有效的保留,因此也被广泛地应用于磷酸化蛋白质组学研究中[47,48]。其主要优势是可以对多磷酸化肽、单磷酸化肽和非磷酸化肽实现更有效的分离。然而,离子交换色谱模式进行多肽分离一般都需要使用高浓度盐进行梯度洗脱,从而引入了额外的柱后除盐步骤,导致样品损失。
3.2 基于高pH反相液相色谱的分离
多肽在反相色谱中的分离是基于其与固定相上的C18基团之间的疏水相互作用实现的。Gilar等[50]首次将高pH反相色谱和低pH反向色谱结合使用,进行二维液相色谱分离,获得了优秀的正交性。该二维分离模式正交性的主要原因可能是肽段在不同的pH条件下电荷分布不同。中科院大连化物所邹汉法研究组对该方法在磷酸化多肽的分离方面进行了系统优化,并创造性地引入了等间隔先流出组分和后流出组分样品混合法(图3)[49]。该方法的优势是:(1)去除了额外的除盐步骤,从而大大降低了微量多肽样品的损失; (2)通过合并样品可以大大减少质谱分析时间,但同时又不降低二维色谱分离的正交性。正是由于上述显著的样品处理优势,该分离策略成为目前蛋白质组学领域中最为广泛使用的多维色谱分离模式[32,51]。基于上述介绍的多维色谱模式, Ficarro等[52]进一步发展了一种基于高pH反相色谱,SAX色谱和低pH反相色谱的三维色谱分离系统; 通过与质谱进行在线联用,最大化地实现了复杂磷酸化多肽样品的分离和高灵敏度质谱检测。作者从1×106~3×106个细胞中(约5~10 μg)共鉴定了约2500个非冗余的磷酸化多肽,是目前应用于蛋白质翻译后修饰研究最灵敏的LC-MS技术。
4 生物质谱鉴定
应用于蛋白质翻译后修饰的生物质谱技术主要需要完成以下任务:(1)鉴定发生修饰的蛋白质; (2)鉴定发生修饰的氨基酸位点及修饰基团的化学结构; (3)定量分析修饰蛋白占总体蛋白表达量的比例。存在于蛋白质上的修饰基团常具有化学稳定性差和丰度低的特点,因此对于质谱检测提出了更高的要求。近年来,多种温和的碎裂技术相继诞生,大大促进了修饰多肽的质谱检测效果。以研究最为广泛的磷酸化修饰为例,本文阐述应用于修饰多肽检测的重要质谱碎裂技术,主要包括:碰撞诱导解离(Collision-induced dissociation, CID)、高能碰撞解离技术(High-energy collisional dissociation,HCD)、基于电子诱导的电子捕获解离(Electron capture dissociation,ECD)和电子转移解离技术(Electron transfer dissociation,ETD)。
4.1 碰撞诱导解离(CID)
CID是串联质谱法中最常用的碰撞解离方法。它利用惰性气体分子撞击肽段母离子使其在酰胺键处断裂,产生具有丰富序列信息的b-和y-型离子。由于磷酸基团在CID过程中易发生中性丢失,影响了它对磷酸化多肽序列的有效鉴定。中性丢失的程度与很多因素有关,如电荷态、化学结构、质子化试剂、碎裂能量等。酪氨酸磷酸化易丢失HPO3(80 Da)的中性碎片,而丝氨酸和苏氨酸磷酸化更倾向于丢失H3PO4 (98 Da)的中性碎片[53]。这主要是由于酪氨酸磷酸化的CO键键能更大,不易碎裂。
为了解决磷酸化肽易发生中性丢失的问题,在离子阱质谱中常进一步采用第三级质谱MS3碎裂,以获得更多的碎片离子信息[54]。多级活化解离,又称虚拟的MS3,将发生中性丢失的产物离子和母离子同时活化,并记录所有的碎片离子[55]。MSA方法常用于低分辨的离子阱质谱中,而在高分辨、高质量精度类质谱(如Orbitrap)中的应用不多[54]。这主要是因为这类质谱可以高通量地获得母离子的精确质量数,因此常规的MS2碎裂已经可以满足检测需求,额外的MS3和多级活化解离反而会消耗本可以用于测序新肽段的质谱时间。
4.2 高能碰撞诱导解离(HCD)
HCD通过在高能碰撞室中提供比 CID更高的碰撞能量和更短的活化时间来解离离子[56,57]。与基于离子阱的CID相比,HCD所产生的中性丢失峰较少,主要产生序列特异性的y-型离子和某些b-型离子(图4)。在HCD中,b-型离子倾向于进一步解离成a-型离子,因此b-型离子的数目较y-型离子少。同时,由于HCD的能量更大,它常产生更小的碎片离子,如磷酸化酪氨酸特异性的Immonium离子。HCD碎裂克服了常规离子阱存在的1/3低质量范围限制,同时提供了比CID更高的分辨率,是Orbitrap质谱仪的标配解离模式,被广泛用于蛋白质翻译后修饰的研究。
4.3 基于电子诱导的解离(ECD/ETD)
与CID相比,基于电子诱导的解离方法,电子捕获解离(ECD)和电子转移解离(ETD),是一种温和的软解离方式,能够产生肽骨架碎裂信息并保留修饰基团。在ECD过程中,母离子被近热电子活化,酰胺键上的氧原子从邻近的氨基酸残基上转移一个质子,此过程放热使N-Cα键发生断裂产生c-型和z-型离子[58]。在ETD过程中,荧蒽自由基阴离子携带电子与肽段阳离子发生相互作用,此过程使多肽电荷数降低并诱导肽骨架的碎裂,产生一系列互补的c-型和z-型离子[59]。与CID相比,ETD更适合高电荷态和包含碱性残基肽段的测序。CID和ETD的互补使用有利于增加蛋白的鉴定数目和提高序列覆盖度,对大规模鉴定磷酸化肽十分有利[60]。另外,ECD和ETD方法是目前应用于糖基化多肽糖链结构解析最为有效的裂解方法[61]。基于上述的解离方法,Heck研究小组发展了一种集成化的解离技术,名为EThcD。顾名思义,该方法将适合磷酸化位点分析的ETD和HCD碎裂技术结合起来[62]。ETD不能断裂N-端连接脯氨酸残基的NCα键,这限制了它对脯氨酸含量高的磷酸化肽的修饰位点的确认。而EThcD方法很好地克服了这方面的缺陷,能够产生丰富的b/y和c/z型离子。
5 结论与展望
蛋白质翻译后修饰多达300多种,它们具有化学稳定性差、丰度低和动态变化等特点,在各种生物学过程中发挥着重要的调控作用。基于LC-MS的蛋白质组学技术已经实现了对成千上万种修饰位点的大规模定性和定量分析,这其中主要包括磷酸化、糖基化、泛素化、乙酰化和甲基化修饰。如本文所述,对这些重要翻译后修饰的大规模蛋白质组学分析的实现主要归功于众多方法学的巨大进步,主要包括高选择性和高效富集方法的发展、高容量和高正交性多维色谱技术的发展以及高灵敏度和高扫描速度生物质谱技术的发展。随着通用型多维色谱技术和生物质谱技术的不断完善,新的蛋白质翻译后修饰的大规模蛋白质组学研究更多局限于高选择性和高效富集方法的发展。如表1所示,在目前主要研究的蛋白质翻译后修饰中,甲基化修饰的大规模质谱鉴定仍处于较低的水平。这主要是因为各种甲基化修饰基团的分子尺寸较小,很难发展高特异性的富集方法。在传统抗体技术无法很好地解决这一难题的情况下,基于MBT甲基化蛋白质结合结构域的富集方法为甲基化蛋白质组研究提供了新颖的高效富集方法[39]。由于几乎所有的重要蛋白质翻译后修饰都有众多与之对应的蛋白质结合结构域,可以预期该类富集方法将会成为未来蛋白质翻译后修饰富集方法发展的重要方向。
此外,随着多种重要蛋白质翻译后修饰大规模蛋白质组学分析方法的逐步成熟,越来越多的研究者开始关注在相关生物体系中的应用研究,尤其是对多种翻译后修饰在同一蛋白质上的协同作用研究。目前的相关研究主要针对大规模质谱鉴定效果最好的翻译后修饰,即磷酸化、泛素化和乙酰化。可以预期,随着其它相关蛋白质翻译后修饰富集方法的不断发展和完善,针对特定生物学体系的多种翻译后修饰的协同作用研究将会得到进一步发展,并在相关生物学研究中起到关键作用。总之,色谱与质谱联用的技术已经为蛋白质翻译后修饰研究提供了革命性的系统水平研究工具,传统生物学领域的“一个基因,一个蛋白质,一种生物学功能”的理念已经被完全打破。基于色谱与质谱联用技术的蛋白质组学策略所产生的海量质谱数据, 已经推动蛋白质翻译后修饰及相关生物学领域进入了全新的系统生物学研究时代。
References
1 Scott J D, Pawson T. Science, 2009, 326: 1220-1224
2 Witze E S, Old W M, Resing K A, Ahn N G. Nat.Methods, 2007, 4: 798-806
3 Choudhary C, Mann M. Nat. Rev. Mol. Cell. Biol., 2010, 11: 427-439
4 Altelaar A F, Munoz J, Heck A J. Nat. Rev. Genet., 2013, 14: 35-48
5 Nilsson C L. Anal. Chem., 2012, 84: 735-746
6 Wang F, Song C, Cheng K, Jiang X, Ye M, Zou H. Anal. Chem., 2011, 83: 8078-8085
7 Sharma K, D′Souza R C, Tyanova S, Schaab C, Wisniewski J R, Cox J, Mann M. Cell Reports, 2014, 8: 1583-1594
8 Udeshi N D, Svinkina T, Mertins P, Kuhn E, Mani D R, Qiao J W, Carr S A. Mol. Cell. Proteomics, 2013, 12: 825-831
9 Lundby A, Lage K, Weinert B T, Bekker-Jensen D B, Secher A, Skovgaard T, Kelstrup C D, Dmytriyev A, Choudhary C, Lundby C, Olsen J V. Cell Reports, 2012, 2: 419-431
10 Guo A, Gu H, Zhou J, Mulhern D, Wang Y, Lee K A, Yang V, Aguiar M, Kornhauser J, Jia X, Ren J, Beausolei S A, Silva J C, Vemulapalli V, Bedford M T, Comb M J. Mol. Cell. Proteomics, 2014, 13: 372-387
11 Zielinska D F, Gnad F, Wisniewski J R, Mann M. Cell, 2010, 141: 897-907
12 Olsen J V, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Cell, 2006, 127: 635-648
13 Sugiyama N, Masuda T, Shinoda K, Nakamura A, Tomita M, Ishihama Y. Mol. Cell. Proteomics, 2007, 6: 1103-1109
14 Ficarro S B, Parikh J R, Blank N C, Marto J A. Anal.Chem., 2008, 80: 4606-4613
15 Larsen M R, Thingholm T E, Jensen O N, Roepstorff P, Jrgensen T J D. Mol. Cell. Proteomics, 2005, 4: 873-886
16 Bodenmiller B, Mueller L N, Mueller M, Domon B, Aebersold R. Nat. Methods, 2007, 4: 231-237
17 Wu J, Shakey Q, Liu W, Schuller A, Follettie M T. J. Proteome Res., 2007, 6: 4684-4689
18 Villen J, Gygi S P. Nat. Protocols, 2008, 3: 1630-1638
19 Zhou H, Ye M, Dong J, Corradini E, Cristobal A, Heck A J, Zou H F, Mohammed S. Nat. Protocols, 2013, 8: 461-480
20 Feng S, Ye M L, Zhou H J, Jiang X G, Jiang X N, Zou H F,Gong B. Mol. Cell. Proteomics, 2007, 6: 1656-1665
21 Thingholm T E, Jensen O N, Robinson P J, Larsen M R. Mol. Cell. Proteomics, 2008, 7: 661-671
22 Zhou H, Ye M, Dong J, Han G, Jiang X, Wu R, Zou H F. J.Proteome Res., 2008, 7: 3957-3967
23 Hunter T, Sefton B M. Proc. Natl. Acad. Sci. U. S. A., 1980, 77: 1311-1315
24 Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H. Cell, 2007, 131: 1190-1203
25 Jorgensen C, Sherman A, Chen G I, Pasculescu A, Poliakov A, Hsiung M, Larsen, B, Wilkinson D G, Linding R, Pawson T. Science, 2009, 326: 1502-1509
26 Tian R, Wang H, Gish G D, Petsalaki E, Pasculescu A, Shi Y, Mollenauerd M, Bagshawa R D, Yoseff N, Huntere T, Gingrasa A C,Weissd A,Pawsona T. Proc. Natl. Acad. Sci. U. S. A., 2015, 112: E1594-E1603
27 Kaji H, Saito H, Yamauchi Y, Shinkawa T, Taoka M, Hirabayashi J, Kasai K, Takahashi N, Isobel T. Nat. Biotechnol., 2003, 21: 667-672
28 Zhang H, Li X J, Martin D B, Aebersold R. Nat. Biotechnol., 2003, 21: 660-666
29 Zhang Y, Yu M, Zhang C, Ma W, Zhang Y, Wang C, Lu H. Anal.Chem., 2014, 86(15): 7920-7924
30 Peng J, Schwartz D, Elias J E, Thoreen C C, Cheng D, Marsischky G, Roelofs J, Finley D, Gygi S P. Nat. Biotech., 2003, 21: 921-926
31 Kim W, Bennett E J, Huttlin E L, Guo A, Li J, Possemato A, Sowa M, Rad R, Rush J, Comb M J, Harper J W, Gygi S P. Mol. Cell, 2011, 44: 325-340
32 Udeshi N D, Mertins P, Svinkina T, Carr SA. Nat.Protocols, 2013, 8: 1950-1960
33 Kim S C, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao Hao, Lin X, Grishin N V,White M, Yang X J, Zhao Y. Mol.Cell, 2006, 23: 607-618
34 Choudhary C, Kumar C, Gnad F, Nielsen M L, Rehman M, Walther T C,Olsen J V, Mann M. Science, 2009, 325: 834-840
35 Boisvert F M, Cté J, Boulanger M C, Richard S. Mol. Cell. Proteomics, 2003, 2: 1319-1330
36 Seet B T, Dikic I, Zhou M M, Pawson T. Nature Reviews Mol. Cell.Biol., 2006, 7: 473-483
37 Machida K, Thompson C M, Dierck K, Jablonowski K, Karkkainen S, Liu B, Zhang H, Nash P D, Newman D K, Nollau P, Pawson T, Renkema G H, Saksela K, Schiller M R, Shin D G, Mayer B J. Mol.Cell, 2007, 26: 899-915
38 Chen C, Nott T J, Jin J, Pawson T. Nat. Rev. Mol.Cell.Biol., 2011, 12: 629-642
39 Moore K E, Carlson S M, Camp N D, Cheung P, James R G, Chua K F,Wolf-Yadlin A, Gozani O. Mol. Cell, 2013, 50: 444-456
40 Mertins P, Qiao J W, Patel J, Udeshi N D, Clauser K R, Mani D R, Burgess M W, Gillette M A, JaffeJ D, Carr S A. Nat. Methods, 2013, 10: 634-637
41 Swaney D L, Beltrao P, Starita L, Guo A, Rush J, Fields S, Krogan N J, Villén J. Nat. Methods, 2013, 10: 676-682
42 Link A J, Eng J, Schieltz D M, Carmack E, Mize G J, Morris D R, Garvik B M, Yates J R. Nat. Biotechnol., 1999, 17: 676-682
43 Motoyama A, Yates J R, 3rd. Anal. Chem., 2008, 80: 7187-7193
44 Gilar M, Olivova P, Daly A E, Gebler J C. Anal. Chem., 2005, 77: 6426-6434
45 Boersema P J, Mohammed S, Heck A J. Anal. Bioanal. Chem., 2008, 391: 151-159
46 Huttlin E L, Jedrychowski M P, Elias J E, Goswami T, Rad R, Beausoleil S A, Villén J, Haas W, Sowa M E, Gygi S P. Cell, 2010, 143: 1174-1189
47 Han G, Ye M, Zhou H, Jiang X, Feng S, Jiang X, Tian R, Wan D, Zou H, Gu J. Proteomics, 2008, 8: 1346-1361
48 Wisniewski J R, Nagaraj N, Zougman A, Gnad F, Mann M. J. Proteome Res., 2010, 9: 3280-3289
49 Song C X, Ye M L, Han G H, Jiang X N, Wang F J, Yu Z Y, Zou H. Anal. Chem., 2010, 82(1): 53-56
50 Gilar M, Olivova P, Daly A E, Gebler J C. J. Sep. Sci., 2005, 28: 1694-1703
51 Kim M S, Pinto S M, Getnet D, Nirujogi R S, Manda S S, Chaerkady R. Nature, 2014, 509: 575-581
52 Ficarro S B, Zhang Y, Carrasco-Alfonso M J, Garg B, Adelmant G, Webber J T. Mol. Cell. Proteomics, 2011, 10: O111 011064
53 Boersema P J, Mohammed S, Heck A J R. J. Mass Spec., 2009, 44: 861-878
54 Villen J, Beausoleil S A, Gygi S P. Proteomics, 2008, 8: 4444-4452
55 Schroeder M J, Shabanowitz J, Schwartz J C, Hunt D F, Coon J J. Anal. Chem., 2004, 76(13): 3590-3598
56 Olsen J V, Macek B, Lange O, Makarov A, Horning S, Mann M. Nat. Methods, 2007, 4: 709-712
57 Jedrychowski M P, Huttlin E L, Haas W, Sowa M E, Rad R, Gygi S P. Mol. Cell. Proteomics, 2011, 10: M111 009910
58 Syrstad E A, Turecek F. J. Am. Soc. Mass Spec., 2005, 16: 208-224
59 Chi A, Huttenhower C, Geer L Y, Coon J J, Syka J E P, Bai D L. Proc. Natl. Acad. Sci. U. S. A., 2007, 104: 2193-2198
60 Phanstiel D H, Brumbaugh J, Wenger C D, Tian S, Probasco M D, Bailey D J. Nat. Methods, 2011, 8: 821-827
61 Zhu Z, Su X, Clark D F, Go E P, Desaire H. Anal. Chem., 2013, 85: 8403-8411
62 Frese C K, Zhou H, Taus T, Altelaar A F M, Mechter K, Heck A J R. J.Proteome Res., 2013, 12: 1520-1525
