基于偶氮苯的光开关分子探针与传感芯片研究进展钦传光
鲁彩霞等
摘 要 随着人们对偶氮苯光致异构化反应机制和特性的研究和认识越来越深入透彻, 偶氮苯衍生物无论是在理论上还是在实验方面都受到了大量的关注。近年来, 偶氮苯衍生物作为光开关响应的功能元件不仅已经用于合成智能聚合物, 液晶材料, 分子开关和分子机器等, 而且正迅速地渗透到化学生物学体系研究和分析的各个方面。因此, 本文将着重就基于偶氮苯衍生物功能化的光开关型分子探针与传感芯片及其在化学生物学分析研究方面所取得的最新进展进行总结和综述。引用103篇文献, 并对未来的发展前景进行了展望。
关键词 偶氮苯衍生物; 分子探针; 传感芯片; 生物分析; 光开关; 分子识别; 综述
1 引 言
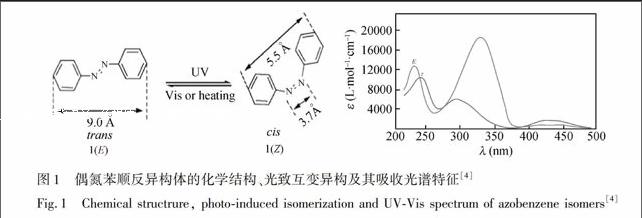
偶氮苯类化合物的发现可以追溯到19世纪, 迄今已经成为化学化工、食品、医药和轻工等诸多行业中一类举足轻重的显色剂、中间体、染料或着色剂[1~3]。偶氮苯1存在两个同分异构体: 反式(Trans)异构1(E)和顺式(Cis)异构体1(Z)。不同的空间排列导致不同的物理和化学性质。偶氮苯的光致异构化反应诱发偶极矩的急剧变化(μ反式偶氮苯 = 0.5 D, 而 μ顺式偶氮苯 = 3.1 D), 进而决定了E和Z同分异构体疏水和亲水特性。反式偶氮苯1(E)不是平面结构, 其二面夹角NNCC大约是17.5°, 而顺式偶氮苯1(Z)中一个苯环所占据的平面与另一苯环的平面呈56°夹角。因此, 偶氮苯的顺、反异构体中4和4′位置上的两个碳原子之间的距离分别是9.0和5.0 。反式偶氮苯1(E)的紫外可见吸收光谱特征由3个主要的带组成: (1)在228 nm处的带源于在苯基上定域的ππ*跃迁; (2) 在318 nm处的带源于在包括两个氮原子的整个分子上离域的对称允许ππ*跃迁; (3)在440 nm处的带源于发生在中央氮原子上对称禁阻的nπ*跃迁。值得注意的是, 顺式偶氮苯1(Z)的紫外可见吸收光谱相当不同于1(E), 其260 nm的带源自对称允许的ππ*跃迁, 而反式异构体的这个带位于318nm处 [4](图1)。
随着人们对偶氮苯光致异构化反应机制和特性的研究和认识的逐渐深入, 偶氮苯衍生物无论是在理论上还是在实验方面都受到了大量关注。近年来, 偶氮苯衍生物作为光开关响应的功能元件不仅已经用于合成智能聚合物[5~7]、液晶材料[8,9]、分子开关[10]和分子机器[11], 而且正迅速地渗透到化学生物学体系研究和分析的各个方面[12,13]。本文将着重就基于偶氮苯衍生物功能化的光开关分子探针与传感芯片及其在化学生物学研究方面所取得的最新进展进行综述。
2 偶氮苯衍生物功能化光开关分子探针
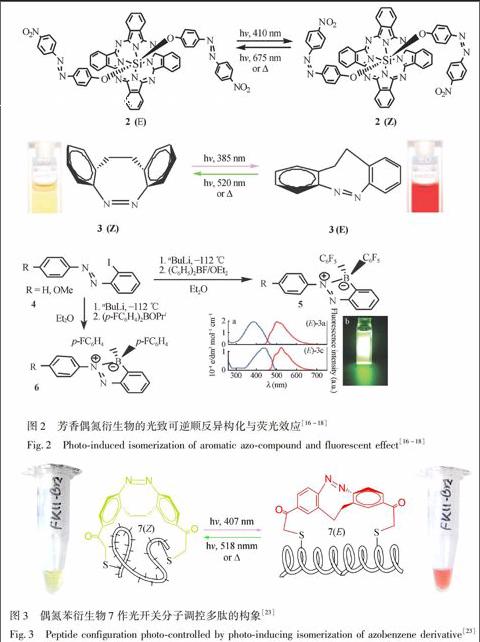
小分子偶氮化合物具有光致异构的特性, 最近被运用于生物系统中作为生物分子(如多肽和蛋白质)的“光开关”, 可以实现对多肽和蛋白质分子构象进行可逆的光化学操作过程, 还可以让多肽和蛋白质等生物分子产生有活性与无活性的转换调控[14,15]。如图2所示, RodríguezRedondo等合成了芳香偶氮衍生物2并观察到其光致顺反异构特性, 用波长410 nm 的光照射反式2(E), 可以转化为其顺式异构体。相反, 若用波长675 nm 的光照射或加热处理顺式2(Z), 则可以转化为其反式异构体2(E)[16]。Siewertsen等研究了桥环偶氮苯3的光致顺反异构特性, 发现其顺式异构体3(Z)(黄色)在385 nm光的照射下可转变为其反式异构体3(E)(红色), 而用520 nm光照或加热处理反式3(E)时, 可以令其恢复到3(Z)的顺式结构[17]。Kawashima研究组采用邻位碘代偶氮苯衍生物4为原料, 合成了一系列的芳香偶氮桥杂环化合物5和6, 并探讨了这些化合的环己烷溶液的荧光激发性质[18]。可见, 芳香偶氮衍生物经功能化后, 可作为光开关, 调控分子和生物荧光成像剂。
在设计合成光控生物分子时, 偶氮化合物开关的光控异构必须考虑以下几个问题:(1)光致异构开关分子必须以某种方式连在生物分子上, 影响生物分子的活性和功能。这取决于作用位点、偶氮开关与多肽或蛋白质相连方式和偶氮衍生物的使用类型等因素。(2)由于顺式和反式异构体的吸收光谱有重叠, 照射产生的光稳定态顺式异构体所占比便最多为80%, 而反式所占比例最多可达95%。热致顺反弛豫, 会产生约100%的异构体。(3)为研究体内光控, 异构化的波长必须与细胞和组织相容。较长波长的光可以较容易地穿透细胞和组织, 且不被细胞内的其它生物大分子所吸收(如NADH)。(4)偶氮衍生物引入细胞后必须是稳定的, 不能改变或退化; 同时, 在探测细胞过程中, 它必须经历多次光开关作用。为探索满足这些要求的新型偶氮光开关分子, 研究者设计合成了一系列偶氮苯衍生物, 并将它们与模式多肽交联, 研究其调控生物分子的构象和活性特征, 筛选并优化偶氮光开关分子结构[19~22]。例如, 以合成的桥环偶氮苯衍生物作为光开关, 交联到α螺旋肽上形成光调控分子7, 研究其光响应特性(图3)[23], 发现7的反式异构体可以稳定多肽的α螺旋构象7(E), 当受到518 nm光照射后, 会异构化而形成顺式的7(Z), 同时, 肽的α螺旋构象遭到破坏而变成无规线状构象, 颜色也由反式的红色变为顺式的黄绿色。而在407 nm光照射下, 又恢复到α螺旋构象7(E)。另外, 也有人合成了一系列的光开关型环肽, 用于研究光对环肽构象的调控变化情况[24,25]。
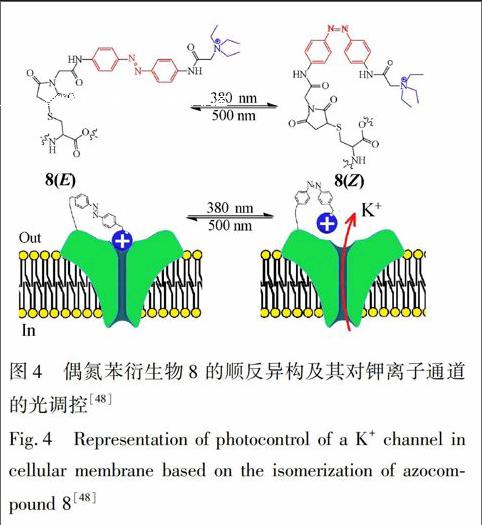
在有生物活性的分子(如蛋白质)中引入偶氮苯分子片段, 能够使各种不同的生物过程通过光照进行时空控制[26~29]。可以通过直接时空调控酶活性[30], 多肽、蛋白质、核酸[25,31,32]、受体[33~38]、离子通道[39~45]等, 或是通过调节几种标记分子的浓度实现对生物过程的时空光控。这个策略是非常有吸引力的, 因为它可以控制生物大分子的构象, 在没有添加任何额外试剂的前提下就能以可逆的方式对生物分子的活性进行调控。由异构化引起的结构效应能够被主体放大或引发一系列的光物理或光化学的二级响应。偶氮苯在生物学中的应用研究发端于上世纪60年代末期, 首次报道其用于光调节胰凝乳蛋白酶(一种消化酶)的活性[46]。后来, 类似的策略被应用于烟碱型乙酰胆碱受体功能和结构的研究[47]。4,4′三甲基铵甲基取代偶氮苯从反式到顺式异构化使乙酰胆碱激动剂的浓度升高, 这是因为两种异构体与存在于兴奋细胞膜上的乙酰胆碱受体具有专一的相互作用。以光致顺反异构互变的开关方式在细胞膜上引起生物电脉冲的过程中允许(或禁阻)离子透膜迁移, 就可能实现光控调节细胞膜渗透能力的改变。偶氮化合物的异构化已经作为合成的控件用来控制细胞膜上离子通道的闭合, 这对离子的跨膜运输而言是很重要的。Trauner等实现了对神经细胞中K+通道的控制就是一个具有说服力的典型例证。偶氮苯衍生物8是一端被固定在细胞膜上的末端季铵盐(图4)[48], 因此, 它处于反式构象8(E)时, 其分子长度让末端季铵盐基团距离正好封堵在钾离子通道上, K+的流动受到阻碍。用波长为380 nm的光照射后, 其转变为顺式异构体8(Z), 使得芳香苯环的距离拉近, 缩短了它的分子长度, 使得末端季铵盐基团的距离无法有效地阻塞K+通道, 允许K+通过。光能够调节离子通道系统的活动, 这在神经生物学中是很重要的。最近, 马来酰亚胺偶氮苯谷氨酸(MAG)9被用作促离子型谷氨酸受体(iGluR)的光致变色激动剂(图5) [49,50], 这个发色团包括一个末端的马来酰亚胺单元, 与蛋白质通过半胱氨酸残基将中心偶氮苯和另一端的谷氨酸共价连接在一起。只有顺式偶氮苯9(Z)才允许谷氨酸片段靠近促离子型谷氨酸受体(iGluR), 谷氨酸此时才能与受体蛋白质的活性位点相互作用。当这种作用发生时, 蛋白质就像贝壳一样折叠起来, 使得离子通道打开。而反式的9(E)则无法拉近谷氨酸与受体蛋白之间的距离, 所以不能有效开启离子通道。利用这种特点, 就可以实现对离子通道进行光控调节。
在多肽中引入了偶氮苯片段, 用于控制α螺旋肽的构象,进而通过这个偶氮光开关来光控调节在生物分子识别中非常重要的构象间相互作用关系。含有两个半胱氨酸残基的多肽可以与分子内含有两个硫醇反应活性基团的偶氮苯光开关进行交联, 偶氮苯的异构化可以通过调节这两个半胱氨酸的空间位置改变多肽的构象。当α螺旋多肽(如DNA识别肽)上交联的偶氮苯基处于反式时, 可维持α螺旋构象并保持原有功能活性(亲和性识别DNA分子)。光照后, 偶氮苯异构化而转变为顺式异构体, 破坏了多肽的螺旋性构象, 就会抑制或使其丧失原有的功能活性(识别DNA的亲和力下降或不能识别DNA了)。若再通过光照使偶氮苯基恢复其反式结构, 则多肽的构象及其功能活性也随之可逆性地恢复。2011年, Woolley研究组在靠近偶氮苯基光开关分子的位置连上荧光染料, 偶氮苯基异构化, 导致荧光变化, 在斑马鱼体内引入被偶氮苯修饰的生物分子, 证明了偶氮苯的光化学在体内与体外是一样的, 适当的偶氮苯衍生物在体内的稳定性可以保持数天[51, 52]。
偶氮苯衍生物作为分子探针的另外一个重要应用是在荧光共振能量转移(FRET)体系中承担荧光淬灭剂的角色。如4(4′二甲氨基苯基偶氮)苯甲酰基(Dabcyl)和4二甲氨基偶氮苯4′磺酰基(Dabsyl)是两种最常见的非荧光受体(可作为荧光淬灭剂), 最大吸收波长分别为 458和 466 nm。其它的偶氮苯衍生物淬灭剂系列还包括 黑洞淬灭剂(Black hole quencher, BHQ)和多路淬灭剂(Multipath quencher, MPQ)[53]。这些淬灭剂有较宽的吸收光谱, 这使它们能作为很多种染料的受体。淬灭剂经常被应用于 DNA 分析, 特别是作为受体和有机染料供体共同构建分子信标[54~56]。这一体系的主要优点是只观测供体分子的信号, 如果光谱范围区分明显, 还可以与其它供体淬灭剂体系共同分析。除了基于 DNA 的诊断, 分子信标还用于测量高分子电解质薄膜对 DNA 的通透性[57], 有催化活性的 DNA 生物传感器还可以检测Pb2+[58]。淬灭剂标记的底物类似物和染料标记的蛋白质被共同用于制备基于 FRET的糖类传感器,可实现对血液检验、食品加工以及发酵制品中糖类化合物的分析与监测[59]。
3 偶氮苯衍生物分子识别的传感芯片
超分子化学是两个或多个化学物种借分子间的弱相互作用力形成的实体或聚集体的化学, 现已成为发展迅速、极富挑战性的新领域。主客体化学是超分子化学的重要分支,其主要研究对象是客体分子(底物)和主体分子(受体)之间的特异性结合,并产生某种特定功能的过程,以及这种结合对两者造成的影响。特异性结合是分子组装及其功能化中的关键过程,也是酶和受体专一选择性的基础。偶氮苯的光致变色特性在“主体客体”分子识别方面同样有应用。例如, 双偶氮化合物10通过氢键间的相互作用作为胍盐离子很好的受体。当偶氮苯为顺式构象10(Z,Z)时, 这种识别是很有效的[60,61]。芳香偶氮衍生物可以作为超分子化学的主体识别并捕获某些有机小分子, 如图6所示, 顺式偶氮苯衍生物10(Z)和11(Z)可以捕获小分子而形成超分子复合物, 受可见光(440 nm)照射, 异构为其反式结构10(E)和11(E)后, 将小分子释放出来[62]。
Pearson等[63]合成了偶氮苯的衍生物12并把它固定在SPR芯片上(图7,左), 实验发现12(E)本身不能与蛋白酶亲和识别, 但紫外光照射芯片后, 12(E)异构化转变为其顺式异构体就能亲和识别蛋白酶了。偶氮苯衍生物的顺式异构体13(Z)通过其两端的寡聚核苷酸片段序列与DNA分子中的互补序列相互识别,能够结合成环状结构。而在可见光照射下,异构化为其反式异构体13(E)后,就只能通过一端的序列与DNA识别,不能形成环状结构(图7,右)。另一方面, 偶氮苯及其衍生物又可以是另一些分子如沸石[64,65]、杯芳烃[66~70]、有机无机超分子通道架构[71]和环糊精[72~74]等的超分子识别对象(或客体) (图8)。特别是芳香偶氮衍生物与环糊精的超分子化学研究开展得相当活跃, 在应用方面也取得了显著进展。
环糊精作为超分子化学主体可以选择性地识别反式偶氮苯及其衍生物, 而不能接纳其顺式异构体。利用这种可逆的超分子识别作用和偶氮化合物的光致异构特性, 人们设计并制备了许多基于偶氮化合物的光敏芯片和智能材料, 用于化学分离、药物递送系统以及生物医学分析。Harada等[75]设计了这样一种单分子层信号输入和读出的超分子系统, β环糊精附于金电极上形成活性界面, 在不同的紫外光照下, 偶氮苯衍生物可与超分子β环糊精进行主客体识别而被包裹或释放, 这样就对信号进行了不同的处理。分子印迹材料是用模板分子在基质表面形成分子印迹所制备的材料,已广泛应用于分离分析和传感器等方面。表面光响应分子印迹材料由于其生色团的存在可光控的摄取或释放分子模板, 这种新型表面印迹系统可通过光控手段提高印迹材料的选择性和分离效率。Wang等[76]设计合成了被偶氮基团功能化的聚醚砜(PES)微纤维, 其中4HA作为分子模板, 它与被偶氮功能化的氨基之间主要的作用是静电引力和氢键作用。在450 nm的光照下, PES结合4HA, 在365 nm的紫外光照下PES释放4HA。聚二甲基硅氧烷(PDMS)表面的功能化得到的材料由于其无毒、易得、良好的光透明性和透气性等优势在生物医学和生物分子领域有很大的应用。尤其是基于PDMS的微流体器件在生物合成、疾病诊断、DNA测序、蛋白质结晶、细胞的生物分析等方面极具吸引力。若将偶氮苯的分子识别从不可逆变得可逆, 就会使得其在生物芯片应用方面显示更为独特的优势。Zhang等[77]利用表面引发原子转移自由基聚合(SIATRP), 通过偶氮苯和环糊精之间的主客体识别相互作用改性PDMS的表面。通过五步反应改性了PDMSPEGCD表面。被罗丹明B标记的偶氮苯和被荧光素FITC标记的偶氮苯与PDMSPEGCD表面在紫外和可见光的照射下进行可逆的组装和分解。首先被标记的反式偶氮苯与表面的PDMSPEGCD在无光下作用12 h, 之后在365 nm的紫外光照射下两者再次分离, 如图9A所示。
在光电传感器件的设计制备方面, 能够识别特异性互补的客体分子的智能纳米管已广受关注, 并且在主客体化学研究领域掀起了研究热潮。Banerjee等[78]研究并探讨了偶氮苯修饰的纳米管和αCD与金层结合的自组装单层膜(αCDSAM)之间的主客体相互作用。首先合成偶氮苯纳米管:羟基偶氮苯羧酸(HABA)上的羧基和多肽纳米管中的氨基之间通过氢键作用, 在4 ℃避光反应48 h后即可制得。制备αCDSAM:将硫醇化的αCD自组装于金层上形成αCDSAM, 反式偶氮苯纳米管因被αCDSAM识别而固定在金的表面。在360 nm的紫外光照5 h, 转变成了顺式偶氮苯纳米管, 就会从αCDSAM上分离, 即顺式偶氮苯结构使得偶氮αCD复合物分解, 纳米管从基质表面移去。这是一个可逆过程, 当在无光下照射24 h,偶氮苯纳米管通过主客体识别作用与αCDSAM又会重新结合, 使得传感芯片表现出智能性, 如图9B所示。
这些智能纳米管能够有效阻止蛋白质不可逆地粘附在其表面的材料, 在生物传感、生物医学移植、体外诊断以及靶向药物递送的载体等方面都有广泛应用。在多种抗蛋白质粘附的材料中, 基于PEG的材料几十年来都被用于阻抗非特异性蛋白质的吸附或细胞粘附。然而, 要想设计出具有反应活性的可固定的生物界面, 使得其能够对外界刺激产生响应, 并可逆地阻止蛋白质吸附, 这依然是一个挑战性的问题。Wan等[79]致力于将该过程进行可逆化控制, 他们利用含偶氮苯的自组装单体(Azo SAM)和pH值响应的嵌段共聚物之间的主客体相互作用, 设计合成了pH值响应的反应活性生物界面。其中pH值响应的嵌段共聚物(PEGPAAgCD)是通过在PEG丙烯酸嵌段共聚物上接枝β环糊精制备的, 再通过材料表面固定的偶氮苯与含环糊精的PEGPAAgCD之间主客体识别相互作用进行自组装, 即形成了PEGPAAgCD的PH响应生物界面。随着pH值变化, 该界面能在延展态和弛豫态之间进行转换, 前者处于pH=7的负电状态, 结合细胞色素c, 后者处于pH=4的电中性, 能阻止界面对细胞色素的吸附, 该过程实现了可逆化, 如图9C所示。
在组织工程研究中, 细胞粘附和迁移是最基本的细胞行为, 其调控过程是可以通过细胞膜上的整合蛋白与结合在聚合物支架上一些特异性生物分子之间的非共价相互作用实现的, 这些特异性生物分子是指诸如层粘连蛋白、纤维连接蛋白和含精氨酸甘氨酸天冬氨酸(RGD)序列的多肽。为了改善聚合物支架对特定细胞的识别性粘附, 人们尝试了各种方法开发仿生生物界面, 其中自组装单层膜(SAMs)技术可以有效地在材料表面引入细胞粘附因子。Gong等[80]利用偶氮苯的光致异构特性在材料表面构筑了基于自组装单层膜(SAM)的“智能芯片”。“智能芯片”是由末端连接链状烷基硅烷的αCD与偶氮苯甘氨酸精氨酸天冬氨酸丝氨酸(azoGRGDS)通过偶氮苯和环糊精的主客体识别进行自组装形成的, 由于存在RGD序列, 这种智能表面可粘附细胞。在加热或可见光照射下, 偶氮苯会处于反式构象, 粘附有细胞的偶氮苯GRGDS就与芯片表面上的αCD发生自组装, 这样就能把细胞固定在芯片表面了。而在紫外光的照射下, 则转变为顺式偶氮苯GRGDS细胞的部分就会脱离芯片表面上的αCDSAMs, 而且这个过程可实现可逆化, 如图9D所示。
最近, Frasconi等[81]利用偶氮苯的光致互变异构效应及其顺反异构体对环糊精选择性主客体识别作用, 在金纳米粒表面构建了固定二茂铁的分子微阵列传感芯片, 通过电化学氧化还原控制组装和逻辑门操控, 实现了对溶液中环糊精接枝聚合物捕获与释放的可逆控制, 用于分离和纯化溶液中的偶氮苯衍生物, 这在水的净化处理和靶向递药方面有潜在的应用价值。此外, 利用偶氮苯环糊精主客体识别体系, 人们还开发出了其它光控智能材料[82]。
4 结论与展望
偶氮苯在上世纪就已为人所知, 研究者对其性能进行了深入的研究。20世纪70年代它已经被应用于生物领域, 但直到最近人们才注意到偶氮苯光控开关有对生物体进行光控的潜力。目前为止主要的挑战依然是以下两个方面:(1)如何使长波开关体现出较大构象变化, 同时维持其独特的开关性能(2)如何转化偶氮苯的光致异构化过程, 使生物分子具有较大的结构和功能转变。虽然, 对偶氮苯衍生物的理化性质和生物学特性进行了较为广泛的研究, 但其分子过程依然不为人们所熟知。偶氮苯光开关的使用为化学生物学的分析方法提供了新思路, 分析并解释了一些生物与生理学问题, 但在化学生物学领域的分析和检测方面, 其应用潜力还有待于深入研究和开发。
目前, 大多数偶氮苯光控开关分子探针和传感芯片的应用还很少涉及对体内生物分子功能的光控。为了进行生物学功能的体内光控实验, 人们既要用光开关修饰特定的生物分子, 引入生物体内(如使用显微注射技术或是膜穿透肽序列)[91~94], 又要有选择性的靶分子。Trauner等使用后面的方法定向研究细胞膜胞外域上的离子通道[95,96], 认为这种离子通道的选择性是由特定的配体/蛋白间相互作用而提供的。使用偶氮苯光开关对细胞内特定靶分子进行修饰时,要求细胞膜必须对这种开头分子具有通透性, 如果对靶分子进行共价修饰, 还需要它能够进行生物正交反应[97,98]。
当然, 为在体内发挥作用, 偶氮苯光开关在还原性的细胞内环境中必须有相当的化学稳定性。细胞内的氧化还原电位由谷胱甘肽(三肽)维持, 其浓度为1~10 mmol/L[99]。该还原机制还涉及到偶氮双键上谷胱甘肽巯基的进攻所形成的甲硫基酰肼的加合物。此类物质能与第二个谷胱甘肽分子反应氧化谷胱甘肽形成非光开关性的肼类化合物。该反应的速率取决于谷胱甘肽的浓度, 修饰后的生物分子浓度以及偶氮苯还原电位。
偶氮苯光开关的其它现象表明, 减少以上条件可以使其在细胞内部更为稳定。Woolley等在体外模拟生理条件下, 在一些氨基偶氮苯中加入较高浓度的谷胱甘肽, 孵育一夜, 并没有使其含量减少[100]。在大肠杆菌代谢活性蛋白中加入苯基偶氮苯丙氨酸可以光控抑制反义密码子的形成。修饰蛋白的成功制备说明了在体内表达过程中含有苯基偶氮苯丙氨酸的多肽(PAP)是较为稳定的。Zhang等[101]研究了光开关性环孢素A的翻译过程, 它含有二氨基偶氮苯基开关, 对谷胱甘肽的还原具有耐受性, 将其溶解在人体血液中可以进行光致异构过程。当然, 除了谷胱甘肽介导的还原可以导致偶氮苯光控开关的减少外, 还有其它路径。酶介导的还原和其它修饰均有多种偶氮染料参与[102,103]。酶介导还原不仅取决于偶氮苯衍生物的氧化还原能力, 还与苯环上的取代基有关。许多证据表明, 偶氮苯光开关性在体内是可以持续的, 因为在某种情况下偶氮苯光开关可以改变细胞的功能。
Woolley等[102]将荧光信号直接加入偶氮苯光开关中以便在体内直接读出偶氮苯光开关。他们制备了一种荧光指示体系, 他们将二氨基偶氮苯光开关交联到一个N末端含有荧光的α螺旋肽。UV照射引起反式顺式异构体转变可以导致荧光强度随时间而减少。蓝光照射引起顺式反式异构体转变可以导致荧光强度随时间而增加。将这种结构注射到斑马鱼的胚胎中, 观察整个生物体(即在各种不同类型的细胞中), 其光开关可以持续至少2天。使用与体外相同的方法测量开关速率、量子产率和光稳定性。以上结果表明, 选择适当的偶氮苯衍生物可探测体内生理过程。例如, 光开关性多肽和蛋白能够探测斑马鱼早期发育的生理模式。这些研究工作为采用偶氮苯光控开关分子探针和传感芯片直接监测或实时调控生物体内的生物学过程奠定了基础并开拓了方向, 必将促进分析化学与生物分析方法学的创新与发展。
References
1 Peters M V, Stoll R S, Kuhn A, Hecht S J. Angew. Chem. Int. Ed., 2008, 47: 5968-5972
2 Wegner A H. Angew. Chem. Int. Ed., 2012, 51: 4787-4788
3 QIN ChuanGuang, LI Yang, LI HaiLiang, LI DaWei, NIU WeiNing, SHANG XiaoYa, XU ChuanLan. Chin. J. Org. Chem., 2013, 33: 444-457
钦传光, 李 洋, 李海亮, 李大为, 牛卫宁, 尚晓娅, 徐春兰. 有机化学, 2013, 33: 444-457
4 Hamon F, DjedainiPilard F, Barbot F, Len C. Tetrahedron, 2009, 65: 10105-10123
5 Russew M M, Hecht S. Adv. Mater., 2010, 22: 3348-3360
6 Feng W, Luo W, Feng Y Y. Nanoscale, 2012, 4: 6118-6134
7 Blasco E, Schmidt B V K J, BarnerKowollik C, Piňol M, Oriol L. Macromol., 2014, 47: 3693-3700
8 Zhang YQ, Ortega J, Baumeister U, Folcia C L, SanzEnguita G, Walker C, RodriguezConde S, Etxebarria J, O′Callaghan M J, More K. J. Am. Chem. Soc., 2012, 134(39): 16298-16306
9 Bobrovsky A, Shibaev V, Cigl M, Hamplov V, Hampl F, Elyashevitch G. J. Mater. Chem. C, 2014, 2: 4482-4489
10 Merino E, Ribagorda M. Beilstein J. Org. Chem., 2012, 8: 1071-1090
11 Sun X M, Wang W, Qiu L B, Guo W H, Yu Y L, Peng H S. Angew. Chem. Int. Ed., 2012, 51: 8520-8524
12 Gorostiza P, Isacoff E Y. Science, 2008, 322: 395-399
13 Wyart C, Del Bene F, Warp E, Scott E K, Trauner D, Baier H, Isacoff E Y. Nature, 2009, 461: 407-410
14 Hoppmann C, Seedorff S, Richter A, Fabian H, Schmieder P. Angew. Chem. Int. Ed., 2009, 48: 6636-6639
15 Beharry A A, Woolley G A. Chem. Soc. Rev., 2011, 40: 4422-4437
16 RodríguezRedondo L J, SastreSantos , FernándezLázaro F, Soares D, Azzellini G C, Elliott B, Echegoyen L. Chem. Commun., 2006, 42: 1265-1268
17 Siewertsen R, Neumann H, BuchheimStehn B, Herges R, Renth F, Temps F. J. Am. Chem. Soc., 2009, 131: 15594-15595
18 Yoshino J, Kano N, Kawashima T. Chem. Commun., 2007, 43: 559-561
19 Beharry A A, Sadovski, O, Woolley G A. J. Am. Chem. Soc., 2011, 133: 19684-19687
20 S amanta S, Woolley G A. ChemBioChem, 2011, 12: 1712-1723
21 Samanta S, McCormick M T, Schmidt K S, Seferos S D, Woolley G A. Chem. Commun., 2013, 49: 10314-10316
22 Samanta S, Babalhavaeji A, Dong M X, Woolley G A. Angew. Chem. Int. Ed., 2013, 52: 14127-14130
23 Samanta S, Qin C G, Lough J A, Woolley G A. Angew. Chem. Int. Ed., 2012, 51: 6452-6455
24 Behrendt R, Schenk M, Musiol HJ G, Moroder L. J. Pep. Sci., 1999, 5: 519-529
25 Renner C, Kusebauch U, Loweneck M, Milbradt A G, Moroder L. J. Pep. Res., 2005, 65: 4-14
26 LI YuChuan, LI ShengHua, QI Cai, ZHANG HuiJuan, ZHU MengYu, PANG SiPing. Acta Chim. Sin., 2011, 69: 2159-2165
李玉川, 李生华, 祁 才, 张慧娟, 朱梦宇, 庞思平. 化学学报, 2011, 69: 2159-2165
27 Schrader T E, Schreier W J, Cordes T, Koller F O, Babitzki G, Denschlag R, Renner C, Lweneck M, Dong S L, Moroder L, Tavan P, Zinth W. Proc. Natl. Acad. Sci. USA. 2007, 104: 15729-15734
28 Asanuma H, Liang X, Nishioka H, Matsunaga D, Liu M, Komiyama M. Nat. Protoc., 2007, 2: 203-212
29 Guerrero L, Smart O S, Weston C J, Burns D C, Woolley G A, Allemann R K. Angew. Chem. Int. Ed., 2005, 44: 7778-7782
30 Nakayama K, Endo M, Majima T. Chem. Commun., 2004, 2386-2387
31 Dong S L, Lweneck M, Schrader T E, Schreier W J, Zinth W, Moroder L, Renner C. Chem. Eur. J., 2006, 12: 1114-1120
32 Woolley G A, Jaikaran A S I, Berezovski M, Calarco J P, Krylov S N, Smart O S, Kumita J R. Biochem., 2006, 45: 6075-6084
33 Pieroni O, Fissi A, Angelini N, Lenci F. Acc. Chem. Res., 2001, 34: 9-17
34 Renner C, Moroder L. ChemBioChem., 2006, 7: 868-878
35 Aaemissegger A, Krutler V, van Gunsteren W F, Hilvert D. J. Am. Chem. Soc., 2005, 127: 2929-2936
36 Sadovski O, Beharry A A, Zhang F, Woolley G A. Angew. Chem. Int. Ed., 2009, 48: 1484-1486
37 Beharry A A, Sadovski O, Woolley G A. J. Am. Chem. Soc., 2011, 133: 19684-19687
38 Ali M A, Woolley G A. Org. Biomol. Chem., 2013, 11: 5325-5331
39 Stawski P, Sumser M, Trauner D. Angew. Chem. Int. Ed., 2012, 51: 5748-5751
40 Tochitsky I, Banghart M R, Mourot A, Yao J Z, Gaub B, Kramer R H, Trauner D. Nat. Chem., 2012, 4: 105-111
41 Stawski P, Janovjak H, Trauner D. Bioorg. Med. Chem., 2010, 18: 7759-7772
42 Kramer R H, Fortin D L, Trauner D. Curr. Opin. Neurobiol., 2009, 19: 544-552
43 Fehrentz T, Schnberger M, Trauner D. Angew. Chem. Int. Ed., 2011, 50: 12156-12182
44 Mourot A, Kienzler M A, Banghart M R, Fehrentz T, Huber F M E, Stein M, Kramer R H, Trauner D. ACS Chem. Neurosci., 2011, 2: 536-543
45 Hilf R J C, Bertozzi C, Zimmermann I, Reiter A, Trauner D, Dutzler R. Nat. Struct. Mol. Biol., 2010, 17: 1330-1336
46 Banghart M, Borges K, Isacoff E, Trauner D, Kramer R H. Nat. Neurosci., 2004, 7: 1381-1386
47 Volgraf M, Gorostiza P, Numano R, Kramer R H, Isacoff E Y, Trauner D. Nat. Chem. Biol., 2006, 2: 47-52
48 Gorostiza P, Volgraf M, Numano R, Szobota S, Trauner D, Isacoff E Y. Proc. Natl. Acad. Sci. USA, 2007, 104: 10865-10870
49 Volgraf M, Gorostiza P, Szobota S, Helix M R, Isacoff E Y, Trauner D. J. Am. Chem. Soc., 2007, 129: 260-261
50 Bredenbeck J, Helbing J, Kumita J R, Woolley G A, Hamm P. Proc. Natl. Acad. Sci. U S A., 2005, 102: 2379-2384
51 Guerrero L, Smart O S, Woolley G.A, Allemann R K. J. Am. Chem. Soc., 2005, 127: 15624-15629
52 Beharry A A, Wong L, Tropepe V, Woolley G A. Angew. Chem. Int. Ed., 2011, 50: 1325-1327
53 Crisalli P, Kool T. Bioconjugate Chem., 2011, 22: 2345-2354
54 Didenko V V. Biotechniques, 2001, 31: 1106-1121
55 Tan L, Li Y, Drake J T, Moroz L, Wang K M, Li J, Munteanu A, Yang C Y J, Martinez K, Tan W H. Analyst, 2005, 130: 1002-1005
56 Tan W H, Wang K M, Drake J T. Curr. Opin. Chem. Biol., 2004, 8: 547-553
57 Johnston P R A, Caruso F. J. Am. Chem. Soc., 2005, 127: 10014-10017
58 Liu J W, Lu Y. Anal. Chem., 2003, 75: 6666-6672
59 Medintz I L, Goldman E R, Lassman M E, Mauro J M. Bioconj. Chem., 2003, 14: 909-912
60 Hunter C A, Togrul M, Tomas S. Chem. Commun., 2004, 108-109
61 Goodman A, Breinlinger E, Ober M, Rotello V M. J. Am. Chem. Soc., 2001, 123: 6213-6214
62 Gomy C, Schmitzer R A. Org. Lett. 2007, 9(2): 3865-3868
63 Pearson D, Abell A D. Chem. Eur. J., 2010, 16: 6983-6992
64 Stawski P, Sumser M, Trauner D. Angew. Chem. Int. Ed., 2012, 51: 5748-5751
65 Tsuwi J, Berger R, Labat G, Couderc G, Behrnd N R, Ottiger P, Cucinotta F, Schurmann K, Bertoni M, Viani L, Cornil J, ProdiSchwab A, de Cola L, Wubbenhorst M, Hulliger J. J. Phys. Chem. A, 2010, 114(26): 6956-6963
66 Kim H S,Cao T, Pham T, Yoon K B. Chem. Commun., 2012, 48: 4659-4673
67 Yu G C, Han C Y, Zhang Z B, Chen J Z, Yan X Z, Zheng B, Liu S Y, Huang F H. J. Am. Chem. Soc., 2012, 134: 8711-8717
68 Dsouza N R, Nau M W. J. Org. Chem., 2008, 73: 5305-5310
69 Bakirci H, Koner L A, Nau M W. J. Org. Chem., 2005, 70: 9960-9966
70 Liu Y, Wang H, Zhang H Y, Liang P. Chem. Commun., 2004, 2266-2267
71 Zaman M B, Udachin K, Akhtaruzzaman M, Yamashita Y, Ripmeester A J. Chem. Commun., 2002, 2322-2323
72 Tomatsu I, Hashidzume A, Harada A. J. Am. Chem. Soc., 2006, 128: 2226-2227
73 Ferris D P, Zhao YL, Khashab N M, Khatib H A, Stoddart J F, Zink J I. J. Am. Chem. Soc., 2009, 131: 1686-1688
74 Liu Y, Yang ZX, Chen Y. J. Org. Chem., 2008, 73: 5298-5304
75 Harada A. Acc. Chem. Res., 2001, 34: 456-464
76 Wang D S, Zhang X X, Nie S Q, Zhao W F, Lu Y, Sun S D, Zhao C S. Langmuir, 2012, 28: 13284-13293
77 Zhang Y R, Ren L, Tu Q, Wang X Q, Liu R, Li L, Wang J C, Liu W M, Xu J, Wang J Y. Anal. Chem., 2011, 83: 9651-9659
78 Banerjee A I, Yu L T, Matsui H. J. Am. Chem. Soc., 2003, 125: 9542-9543
79 Wan P B, Chen Y Y, Xing Y B, Chi L F, Zhang X. Langmuir, 2010, 26: 12515-12517
80 Gong Y H, Li C, Yang J, Wang H Y, Zhuo R X, Zhang X Z. Macromol., 2011, 44: 7499-7502
81 Frasconi M, Mazzei F. Langmuir, 2012, 28: 3322-3331
82 Tamesue S, Takashima Y, Yamaguchi H, Shinkai S, Harada A. Angew. Chem., 2010, 122: 7623-7626
83 Liu J H, Chen G S, Guo M Y, Jiang M. Macromol. 2010, 43: 8086-8093
84 Liao X J, Chen G S, Liu X X, Chen W X, Chen F E, Jiang M. Angew. Chem. Int. Ed., 2010, 49: 4409-4413
85 Liao X J, Chen G S, Jiang M. Langmuir, 2011, 27: 12650-12656
86 Zhang H J, Xin Y, Yan Q, Zhou L L, Peng L, Yuan J Y. Macromol. Rapid Commun. 2012, 33: 1952-1957
87 Liu R, Zhang Y, Feng P Y. J. Am. Chem. Soc., 2009, 131: 15128-15129
88 Wang D S, Zhang X X, Nie S Q, Zhao W F, Lu Y, Sun S D, Zhao C S. Langmuir 2012, 28: 13284-13293
89 Jin H B, Zheng Y L, Liu Y, Cheng H X, Zhou Y F, Yan D Y. Angew. Chem. Int. Ed., 2011, 50: 10352-10356
90 Wu M, Cao Y P, Zhang X Z, Zhang Y F, Chen Y, He L, Qian Z Y. Chem. Commun., 2012, 48: 9846-9848
91 Woolley, G A. Nat. Chem., 2012, 4: 75-77
92 Woolley, G A. Nat. Nanotechnol., 2013, 8: 892-893
93 QIN ChuanGuang,ZHANG Yuan, LI Lin, SHANG XiaoYa, NIU WeiNing, XU ChuanLan. Sci. Sin. Chim., 2013, 43: 1322-1335
钦传光, 张 媛, 李 琳, 尚晓娅, 牛卫宁, 徐春兰. 中国科学: 化学, 2013, 43: 1322-1335
94 REN Jin, QIN ChuanGuang, XU ChuanLan, WANG QiuYu, ZUO XiaoJia. Act. Pharmceut. Sin., 2010, 45: 17-25
任 锦, 钦传光, 徐春兰, 王秋雨, 左晓佳. 药学学报, 2010, 45: 17-25
95 Zhang F, Timm A K, Arndt M K, Woolley G A. Angew. Chem. Int. Ed., 2010, 49: 3943-3946
96 Szobota S, Isacoff Y E. Annu. Rev. Biophys. Biomol. Struct., 2010, 39: 329-348
97 Banghart R M, Mourot A, Fortin L D, Yao J Z, Kramer R H, Trauner D. Angew. Chem. Int. Ed., 2009, 48: 9097-9101
98 Lim R K, Lin Q. Chem. Commun., 2010, 46: 1589-1600
99 Boulegue C, Loweneck M, Renner C, Moroder L. ChemBioChem, 2007, 8: 591-594
100 Bose M, Groff D, Xie J, Brustad E, Schultz P G. J. Am. Chem. Soc., 2006, 128: 388-389
101 Zhang Y, Erdmann F, Fischer G. Nat. Chem. Biol., 2009, 5: 724-726
102 Sadovski O, Beharry A A, Zhang F, Woolley G A. Angew. Chem. Int. Ed., 2009, 48: 1484-1486
103 Beharry A A, Sadovski O, Woolley G A. Org. Biomol. Chem., 2008, 6: 4323-4332
Abstract Azobenzene derivatives have received considerable experimental and theoretical attention, along with more and more the investigation and the knowledge on the characteristics of their photoinduced isomerization. Except for their traditionally wellknown uses as dyes or colorants in many industries, in recent years, azobenzene derivatives have been widely used as photoresponsive functional devices utilized not only as smart polymers, liquid crystals, molecular switches, and machines, which have been reviewed in several reports. Simultaneously, they also have been rapidly permeated and applied to the field of chemical and biological analysis as photoswitchable molecular probes and sensory chips, playing an increasingly significant role. So, it is worth emphasizing to review and summarize the last developments in the later topics as title of this paper. More than 100 relevant literatures are cited here and the prospects are pointed out.
Keywords Azobenzene derivative; Molecular probe; Sensory chip; Bioanalysis and imaging; Photoswitching agent; Molecular recognition; Review
(Received 8 October 2014; accepted 22 December 2014)
This work was supported by the National Natural Science Foundation of China (Nos. 20672086, 20802057) and the Natural Science Basic Research Plan in Shaanxi Province of China (No. 2012JZ2002)
摘 要 随着人们对偶氮苯光致异构化反应机制和特性的研究和认识越来越深入透彻, 偶氮苯衍生物无论是在理论上还是在实验方面都受到了大量的关注。近年来, 偶氮苯衍生物作为光开关响应的功能元件不仅已经用于合成智能聚合物, 液晶材料, 分子开关和分子机器等, 而且正迅速地渗透到化学生物学体系研究和分析的各个方面。因此, 本文将着重就基于偶氮苯衍生物功能化的光开关型分子探针与传感芯片及其在化学生物学分析研究方面所取得的最新进展进行总结和综述。引用103篇文献, 并对未来的发展前景进行了展望。
关键词 偶氮苯衍生物; 分子探针; 传感芯片; 生物分析; 光开关; 分子识别; 综述
1 引 言
偶氮苯类化合物的发现可以追溯到19世纪, 迄今已经成为化学化工、食品、医药和轻工等诸多行业中一类举足轻重的显色剂、中间体、染料或着色剂[1~3]。偶氮苯1存在两个同分异构体: 反式(Trans)异构1(E)和顺式(Cis)异构体1(Z)。不同的空间排列导致不同的物理和化学性质。偶氮苯的光致异构化反应诱发偶极矩的急剧变化(μ反式偶氮苯 = 0.5 D, 而 μ顺式偶氮苯 = 3.1 D), 进而决定了E和Z同分异构体疏水和亲水特性。反式偶氮苯1(E)不是平面结构, 其二面夹角NNCC大约是17.5°, 而顺式偶氮苯1(Z)中一个苯环所占据的平面与另一苯环的平面呈56°夹角。因此, 偶氮苯的顺、反异构体中4和4′位置上的两个碳原子之间的距离分别是9.0和5.0 。反式偶氮苯1(E)的紫外可见吸收光谱特征由3个主要的带组成: (1)在228 nm处的带源于在苯基上定域的ππ*跃迁; (2) 在318 nm处的带源于在包括两个氮原子的整个分子上离域的对称允许ππ*跃迁; (3)在440 nm处的带源于发生在中央氮原子上对称禁阻的nπ*跃迁。值得注意的是, 顺式偶氮苯1(Z)的紫外可见吸收光谱相当不同于1(E), 其260 nm的带源自对称允许的ππ*跃迁, 而反式异构体的这个带位于318nm处 [4](图1)。
随着人们对偶氮苯光致异构化反应机制和特性的研究和认识的逐渐深入, 偶氮苯衍生物无论是在理论上还是在实验方面都受到了大量关注。近年来, 偶氮苯衍生物作为光开关响应的功能元件不仅已经用于合成智能聚合物[5~7]、液晶材料[8,9]、分子开关[10]和分子机器[11], 而且正迅速地渗透到化学生物学体系研究和分析的各个方面[12,13]。本文将着重就基于偶氮苯衍生物功能化的光开关分子探针与传感芯片及其在化学生物学研究方面所取得的最新进展进行综述。
2 偶氮苯衍生物功能化光开关分子探针
小分子偶氮化合物具有光致异构的特性, 最近被运用于生物系统中作为生物分子(如多肽和蛋白质)的“光开关”, 可以实现对多肽和蛋白质分子构象进行可逆的光化学操作过程, 还可以让多肽和蛋白质等生物分子产生有活性与无活性的转换调控[14,15]。如图2所示, RodríguezRedondo等合成了芳香偶氮衍生物2并观察到其光致顺反异构特性, 用波长410 nm 的光照射反式2(E), 可以转化为其顺式异构体。相反, 若用波长675 nm 的光照射或加热处理顺式2(Z), 则可以转化为其反式异构体2(E)[16]。Siewertsen等研究了桥环偶氮苯3的光致顺反异构特性, 发现其顺式异构体3(Z)(黄色)在385 nm光的照射下可转变为其反式异构体3(E)(红色), 而用520 nm光照或加热处理反式3(E)时, 可以令其恢复到3(Z)的顺式结构[17]。Kawashima研究组采用邻位碘代偶氮苯衍生物4为原料, 合成了一系列的芳香偶氮桥杂环化合物5和6, 并探讨了这些化合的环己烷溶液的荧光激发性质[18]。可见, 芳香偶氮衍生物经功能化后, 可作为光开关, 调控分子和生物荧光成像剂。
在设计合成光控生物分子时, 偶氮化合物开关的光控异构必须考虑以下几个问题:(1)光致异构开关分子必须以某种方式连在生物分子上, 影响生物分子的活性和功能。这取决于作用位点、偶氮开关与多肽或蛋白质相连方式和偶氮衍生物的使用类型等因素。(2)由于顺式和反式异构体的吸收光谱有重叠, 照射产生的光稳定态顺式异构体所占比便最多为80%, 而反式所占比例最多可达95%。热致顺反弛豫, 会产生约100%的异构体。(3)为研究体内光控, 异构化的波长必须与细胞和组织相容。较长波长的光可以较容易地穿透细胞和组织, 且不被细胞内的其它生物大分子所吸收(如NADH)。(4)偶氮衍生物引入细胞后必须是稳定的, 不能改变或退化; 同时, 在探测细胞过程中, 它必须经历多次光开关作用。为探索满足这些要求的新型偶氮光开关分子, 研究者设计合成了一系列偶氮苯衍生物, 并将它们与模式多肽交联, 研究其调控生物分子的构象和活性特征, 筛选并优化偶氮光开关分子结构[19~22]。例如, 以合成的桥环偶氮苯衍生物作为光开关, 交联到α螺旋肽上形成光调控分子7, 研究其光响应特性(图3)[23], 发现7的反式异构体可以稳定多肽的α螺旋构象7(E), 当受到518 nm光照射后, 会异构化而形成顺式的7(Z), 同时, 肽的α螺旋构象遭到破坏而变成无规线状构象, 颜色也由反式的红色变为顺式的黄绿色。而在407 nm光照射下, 又恢复到α螺旋构象7(E)。另外, 也有人合成了一系列的光开关型环肽, 用于研究光对环肽构象的调控变化情况[24,25]。
在有生物活性的分子(如蛋白质)中引入偶氮苯分子片段, 能够使各种不同的生物过程通过光照进行时空控制[26~29]。可以通过直接时空调控酶活性[30], 多肽、蛋白质、核酸[25,31,32]、受体[33~38]、离子通道[39~45]等, 或是通过调节几种标记分子的浓度实现对生物过程的时空光控。这个策略是非常有吸引力的, 因为它可以控制生物大分子的构象, 在没有添加任何额外试剂的前提下就能以可逆的方式对生物分子的活性进行调控。由异构化引起的结构效应能够被主体放大或引发一系列的光物理或光化学的二级响应。偶氮苯在生物学中的应用研究发端于上世纪60年代末期, 首次报道其用于光调节胰凝乳蛋白酶(一种消化酶)的活性[46]。后来, 类似的策略被应用于烟碱型乙酰胆碱受体功能和结构的研究[47]。4,4′三甲基铵甲基取代偶氮苯从反式到顺式异构化使乙酰胆碱激动剂的浓度升高, 这是因为两种异构体与存在于兴奋细胞膜上的乙酰胆碱受体具有专一的相互作用。以光致顺反异构互变的开关方式在细胞膜上引起生物电脉冲的过程中允许(或禁阻)离子透膜迁移, 就可能实现光控调节细胞膜渗透能力的改变。偶氮化合物的异构化已经作为合成的控件用来控制细胞膜上离子通道的闭合, 这对离子的跨膜运输而言是很重要的。Trauner等实现了对神经细胞中K+通道的控制就是一个具有说服力的典型例证。偶氮苯衍生物8是一端被固定在细胞膜上的末端季铵盐(图4)[48], 因此, 它处于反式构象8(E)时, 其分子长度让末端季铵盐基团距离正好封堵在钾离子通道上, K+的流动受到阻碍。用波长为380 nm的光照射后, 其转变为顺式异构体8(Z), 使得芳香苯环的距离拉近, 缩短了它的分子长度, 使得末端季铵盐基团的距离无法有效地阻塞K+通道, 允许K+通过。光能够调节离子通道系统的活动, 这在神经生物学中是很重要的。最近, 马来酰亚胺偶氮苯谷氨酸(MAG)9被用作促离子型谷氨酸受体(iGluR)的光致变色激动剂(图5) [49,50], 这个发色团包括一个末端的马来酰亚胺单元, 与蛋白质通过半胱氨酸残基将中心偶氮苯和另一端的谷氨酸共价连接在一起。只有顺式偶氮苯9(Z)才允许谷氨酸片段靠近促离子型谷氨酸受体(iGluR), 谷氨酸此时才能与受体蛋白质的活性位点相互作用。当这种作用发生时, 蛋白质就像贝壳一样折叠起来, 使得离子通道打开。而反式的9(E)则无法拉近谷氨酸与受体蛋白之间的距离, 所以不能有效开启离子通道。利用这种特点, 就可以实现对离子通道进行光控调节。
在多肽中引入了偶氮苯片段, 用于控制α螺旋肽的构象,进而通过这个偶氮光开关来光控调节在生物分子识别中非常重要的构象间相互作用关系。含有两个半胱氨酸残基的多肽可以与分子内含有两个硫醇反应活性基团的偶氮苯光开关进行交联, 偶氮苯的异构化可以通过调节这两个半胱氨酸的空间位置改变多肽的构象。当α螺旋多肽(如DNA识别肽)上交联的偶氮苯基处于反式时, 可维持α螺旋构象并保持原有功能活性(亲和性识别DNA分子)。光照后, 偶氮苯异构化而转变为顺式异构体, 破坏了多肽的螺旋性构象, 就会抑制或使其丧失原有的功能活性(识别DNA的亲和力下降或不能识别DNA了)。若再通过光照使偶氮苯基恢复其反式结构, 则多肽的构象及其功能活性也随之可逆性地恢复。2011年, Woolley研究组在靠近偶氮苯基光开关分子的位置连上荧光染料, 偶氮苯基异构化, 导致荧光变化, 在斑马鱼体内引入被偶氮苯修饰的生物分子, 证明了偶氮苯的光化学在体内与体外是一样的, 适当的偶氮苯衍生物在体内的稳定性可以保持数天[51, 52]。
偶氮苯衍生物作为分子探针的另外一个重要应用是在荧光共振能量转移(FRET)体系中承担荧光淬灭剂的角色。如4(4′二甲氨基苯基偶氮)苯甲酰基(Dabcyl)和4二甲氨基偶氮苯4′磺酰基(Dabsyl)是两种最常见的非荧光受体(可作为荧光淬灭剂), 最大吸收波长分别为 458和 466 nm。其它的偶氮苯衍生物淬灭剂系列还包括 黑洞淬灭剂(Black hole quencher, BHQ)和多路淬灭剂(Multipath quencher, MPQ)[53]。这些淬灭剂有较宽的吸收光谱, 这使它们能作为很多种染料的受体。淬灭剂经常被应用于 DNA 分析, 特别是作为受体和有机染料供体共同构建分子信标[54~56]。这一体系的主要优点是只观测供体分子的信号, 如果光谱范围区分明显, 还可以与其它供体淬灭剂体系共同分析。除了基于 DNA 的诊断, 分子信标还用于测量高分子电解质薄膜对 DNA 的通透性[57], 有催化活性的 DNA 生物传感器还可以检测Pb2+[58]。淬灭剂标记的底物类似物和染料标记的蛋白质被共同用于制备基于 FRET的糖类传感器,可实现对血液检验、食品加工以及发酵制品中糖类化合物的分析与监测[59]。
3 偶氮苯衍生物分子识别的传感芯片
超分子化学是两个或多个化学物种借分子间的弱相互作用力形成的实体或聚集体的化学, 现已成为发展迅速、极富挑战性的新领域。主客体化学是超分子化学的重要分支,其主要研究对象是客体分子(底物)和主体分子(受体)之间的特异性结合,并产生某种特定功能的过程,以及这种结合对两者造成的影响。特异性结合是分子组装及其功能化中的关键过程,也是酶和受体专一选择性的基础。偶氮苯的光致变色特性在“主体客体”分子识别方面同样有应用。例如, 双偶氮化合物10通过氢键间的相互作用作为胍盐离子很好的受体。当偶氮苯为顺式构象10(Z,Z)时, 这种识别是很有效的[60,61]。芳香偶氮衍生物可以作为超分子化学的主体识别并捕获某些有机小分子, 如图6所示, 顺式偶氮苯衍生物10(Z)和11(Z)可以捕获小分子而形成超分子复合物, 受可见光(440 nm)照射, 异构为其反式结构10(E)和11(E)后, 将小分子释放出来[62]。
Pearson等[63]合成了偶氮苯的衍生物12并把它固定在SPR芯片上(图7,左), 实验发现12(E)本身不能与蛋白酶亲和识别, 但紫外光照射芯片后, 12(E)异构化转变为其顺式异构体就能亲和识别蛋白酶了。偶氮苯衍生物的顺式异构体13(Z)通过其两端的寡聚核苷酸片段序列与DNA分子中的互补序列相互识别,能够结合成环状结构。而在可见光照射下,异构化为其反式异构体13(E)后,就只能通过一端的序列与DNA识别,不能形成环状结构(图7,右)。另一方面, 偶氮苯及其衍生物又可以是另一些分子如沸石[64,65]、杯芳烃[66~70]、有机无机超分子通道架构[71]和环糊精[72~74]等的超分子识别对象(或客体) (图8)。特别是芳香偶氮衍生物与环糊精的超分子化学研究开展得相当活跃, 在应用方面也取得了显著进展。
环糊精作为超分子化学主体可以选择性地识别反式偶氮苯及其衍生物, 而不能接纳其顺式异构体。利用这种可逆的超分子识别作用和偶氮化合物的光致异构特性, 人们设计并制备了许多基于偶氮化合物的光敏芯片和智能材料, 用于化学分离、药物递送系统以及生物医学分析。Harada等[75]设计了这样一种单分子层信号输入和读出的超分子系统, β环糊精附于金电极上形成活性界面, 在不同的紫外光照下, 偶氮苯衍生物可与超分子β环糊精进行主客体识别而被包裹或释放, 这样就对信号进行了不同的处理。分子印迹材料是用模板分子在基质表面形成分子印迹所制备的材料,已广泛应用于分离分析和传感器等方面。表面光响应分子印迹材料由于其生色团的存在可光控的摄取或释放分子模板, 这种新型表面印迹系统可通过光控手段提高印迹材料的选择性和分离效率。Wang等[76]设计合成了被偶氮基团功能化的聚醚砜(PES)微纤维, 其中4HA作为分子模板, 它与被偶氮功能化的氨基之间主要的作用是静电引力和氢键作用。在450 nm的光照下, PES结合4HA, 在365 nm的紫外光照下PES释放4HA。聚二甲基硅氧烷(PDMS)表面的功能化得到的材料由于其无毒、易得、良好的光透明性和透气性等优势在生物医学和生物分子领域有很大的应用。尤其是基于PDMS的微流体器件在生物合成、疾病诊断、DNA测序、蛋白质结晶、细胞的生物分析等方面极具吸引力。若将偶氮苯的分子识别从不可逆变得可逆, 就会使得其在生物芯片应用方面显示更为独特的优势。Zhang等[77]利用表面引发原子转移自由基聚合(SIATRP), 通过偶氮苯和环糊精之间的主客体识别相互作用改性PDMS的表面。通过五步反应改性了PDMSPEGCD表面。被罗丹明B标记的偶氮苯和被荧光素FITC标记的偶氮苯与PDMSPEGCD表面在紫外和可见光的照射下进行可逆的组装和分解。首先被标记的反式偶氮苯与表面的PDMSPEGCD在无光下作用12 h, 之后在365 nm的紫外光照射下两者再次分离, 如图9A所示。
在光电传感器件的设计制备方面, 能够识别特异性互补的客体分子的智能纳米管已广受关注, 并且在主客体化学研究领域掀起了研究热潮。Banerjee等[78]研究并探讨了偶氮苯修饰的纳米管和αCD与金层结合的自组装单层膜(αCDSAM)之间的主客体相互作用。首先合成偶氮苯纳米管:羟基偶氮苯羧酸(HABA)上的羧基和多肽纳米管中的氨基之间通过氢键作用, 在4 ℃避光反应48 h后即可制得。制备αCDSAM:将硫醇化的αCD自组装于金层上形成αCDSAM, 反式偶氮苯纳米管因被αCDSAM识别而固定在金的表面。在360 nm的紫外光照5 h, 转变成了顺式偶氮苯纳米管, 就会从αCDSAM上分离, 即顺式偶氮苯结构使得偶氮αCD复合物分解, 纳米管从基质表面移去。这是一个可逆过程, 当在无光下照射24 h,偶氮苯纳米管通过主客体识别作用与αCDSAM又会重新结合, 使得传感芯片表现出智能性, 如图9B所示。
这些智能纳米管能够有效阻止蛋白质不可逆地粘附在其表面的材料, 在生物传感、生物医学移植、体外诊断以及靶向药物递送的载体等方面都有广泛应用。在多种抗蛋白质粘附的材料中, 基于PEG的材料几十年来都被用于阻抗非特异性蛋白质的吸附或细胞粘附。然而, 要想设计出具有反应活性的可固定的生物界面, 使得其能够对外界刺激产生响应, 并可逆地阻止蛋白质吸附, 这依然是一个挑战性的问题。Wan等[79]致力于将该过程进行可逆化控制, 他们利用含偶氮苯的自组装单体(Azo SAM)和pH值响应的嵌段共聚物之间的主客体相互作用, 设计合成了pH值响应的反应活性生物界面。其中pH值响应的嵌段共聚物(PEGPAAgCD)是通过在PEG丙烯酸嵌段共聚物上接枝β环糊精制备的, 再通过材料表面固定的偶氮苯与含环糊精的PEGPAAgCD之间主客体识别相互作用进行自组装, 即形成了PEGPAAgCD的PH响应生物界面。随着pH值变化, 该界面能在延展态和弛豫态之间进行转换, 前者处于pH=7的负电状态, 结合细胞色素c, 后者处于pH=4的电中性, 能阻止界面对细胞色素的吸附, 该过程实现了可逆化, 如图9C所示。
在组织工程研究中, 细胞粘附和迁移是最基本的细胞行为, 其调控过程是可以通过细胞膜上的整合蛋白与结合在聚合物支架上一些特异性生物分子之间的非共价相互作用实现的, 这些特异性生物分子是指诸如层粘连蛋白、纤维连接蛋白和含精氨酸甘氨酸天冬氨酸(RGD)序列的多肽。为了改善聚合物支架对特定细胞的识别性粘附, 人们尝试了各种方法开发仿生生物界面, 其中自组装单层膜(SAMs)技术可以有效地在材料表面引入细胞粘附因子。Gong等[80]利用偶氮苯的光致异构特性在材料表面构筑了基于自组装单层膜(SAM)的“智能芯片”。“智能芯片”是由末端连接链状烷基硅烷的αCD与偶氮苯甘氨酸精氨酸天冬氨酸丝氨酸(azoGRGDS)通过偶氮苯和环糊精的主客体识别进行自组装形成的, 由于存在RGD序列, 这种智能表面可粘附细胞。在加热或可见光照射下, 偶氮苯会处于反式构象, 粘附有细胞的偶氮苯GRGDS就与芯片表面上的αCD发生自组装, 这样就能把细胞固定在芯片表面了。而在紫外光的照射下, 则转变为顺式偶氮苯GRGDS细胞的部分就会脱离芯片表面上的αCDSAMs, 而且这个过程可实现可逆化, 如图9D所示。
最近, Frasconi等[81]利用偶氮苯的光致互变异构效应及其顺反异构体对环糊精选择性主客体识别作用, 在金纳米粒表面构建了固定二茂铁的分子微阵列传感芯片, 通过电化学氧化还原控制组装和逻辑门操控, 实现了对溶液中环糊精接枝聚合物捕获与释放的可逆控制, 用于分离和纯化溶液中的偶氮苯衍生物, 这在水的净化处理和靶向递药方面有潜在的应用价值。此外, 利用偶氮苯环糊精主客体识别体系, 人们还开发出了其它光控智能材料[82]。
4 结论与展望
偶氮苯在上世纪就已为人所知, 研究者对其性能进行了深入的研究。20世纪70年代它已经被应用于生物领域, 但直到最近人们才注意到偶氮苯光控开关有对生物体进行光控的潜力。目前为止主要的挑战依然是以下两个方面:(1)如何使长波开关体现出较大构象变化, 同时维持其独特的开关性能(2)如何转化偶氮苯的光致异构化过程, 使生物分子具有较大的结构和功能转变。虽然, 对偶氮苯衍生物的理化性质和生物学特性进行了较为广泛的研究, 但其分子过程依然不为人们所熟知。偶氮苯光开关的使用为化学生物学的分析方法提供了新思路, 分析并解释了一些生物与生理学问题, 但在化学生物学领域的分析和检测方面, 其应用潜力还有待于深入研究和开发。
目前, 大多数偶氮苯光控开关分子探针和传感芯片的应用还很少涉及对体内生物分子功能的光控。为了进行生物学功能的体内光控实验, 人们既要用光开关修饰特定的生物分子, 引入生物体内(如使用显微注射技术或是膜穿透肽序列)[91~94], 又要有选择性的靶分子。Trauner等使用后面的方法定向研究细胞膜胞外域上的离子通道[95,96], 认为这种离子通道的选择性是由特定的配体/蛋白间相互作用而提供的。使用偶氮苯光开关对细胞内特定靶分子进行修饰时,要求细胞膜必须对这种开头分子具有通透性, 如果对靶分子进行共价修饰, 还需要它能够进行生物正交反应[97,98]。
当然, 为在体内发挥作用, 偶氮苯光开关在还原性的细胞内环境中必须有相当的化学稳定性。细胞内的氧化还原电位由谷胱甘肽(三肽)维持, 其浓度为1~10 mmol/L[99]。该还原机制还涉及到偶氮双键上谷胱甘肽巯基的进攻所形成的甲硫基酰肼的加合物。此类物质能与第二个谷胱甘肽分子反应氧化谷胱甘肽形成非光开关性的肼类化合物。该反应的速率取决于谷胱甘肽的浓度, 修饰后的生物分子浓度以及偶氮苯还原电位。
偶氮苯光开关的其它现象表明, 减少以上条件可以使其在细胞内部更为稳定。Woolley等在体外模拟生理条件下, 在一些氨基偶氮苯中加入较高浓度的谷胱甘肽, 孵育一夜, 并没有使其含量减少[100]。在大肠杆菌代谢活性蛋白中加入苯基偶氮苯丙氨酸可以光控抑制反义密码子的形成。修饰蛋白的成功制备说明了在体内表达过程中含有苯基偶氮苯丙氨酸的多肽(PAP)是较为稳定的。Zhang等[101]研究了光开关性环孢素A的翻译过程, 它含有二氨基偶氮苯基开关, 对谷胱甘肽的还原具有耐受性, 将其溶解在人体血液中可以进行光致异构过程。当然, 除了谷胱甘肽介导的还原可以导致偶氮苯光控开关的减少外, 还有其它路径。酶介导的还原和其它修饰均有多种偶氮染料参与[102,103]。酶介导还原不仅取决于偶氮苯衍生物的氧化还原能力, 还与苯环上的取代基有关。许多证据表明, 偶氮苯光开关性在体内是可以持续的, 因为在某种情况下偶氮苯光开关可以改变细胞的功能。
Woolley等[102]将荧光信号直接加入偶氮苯光开关中以便在体内直接读出偶氮苯光开关。他们制备了一种荧光指示体系, 他们将二氨基偶氮苯光开关交联到一个N末端含有荧光的α螺旋肽。UV照射引起反式顺式异构体转变可以导致荧光强度随时间而减少。蓝光照射引起顺式反式异构体转变可以导致荧光强度随时间而增加。将这种结构注射到斑马鱼的胚胎中, 观察整个生物体(即在各种不同类型的细胞中), 其光开关可以持续至少2天。使用与体外相同的方法测量开关速率、量子产率和光稳定性。以上结果表明, 选择适当的偶氮苯衍生物可探测体内生理过程。例如, 光开关性多肽和蛋白能够探测斑马鱼早期发育的生理模式。这些研究工作为采用偶氮苯光控开关分子探针和传感芯片直接监测或实时调控生物体内的生物学过程奠定了基础并开拓了方向, 必将促进分析化学与生物分析方法学的创新与发展。
References
1 Peters M V, Stoll R S, Kuhn A, Hecht S J. Angew. Chem. Int. Ed., 2008, 47: 5968-5972
2 Wegner A H. Angew. Chem. Int. Ed., 2012, 51: 4787-4788
3 QIN ChuanGuang, LI Yang, LI HaiLiang, LI DaWei, NIU WeiNing, SHANG XiaoYa, XU ChuanLan. Chin. J. Org. Chem., 2013, 33: 444-457
钦传光, 李 洋, 李海亮, 李大为, 牛卫宁, 尚晓娅, 徐春兰. 有机化学, 2013, 33: 444-457
4 Hamon F, DjedainiPilard F, Barbot F, Len C. Tetrahedron, 2009, 65: 10105-10123
5 Russew M M, Hecht S. Adv. Mater., 2010, 22: 3348-3360
6 Feng W, Luo W, Feng Y Y. Nanoscale, 2012, 4: 6118-6134
7 Blasco E, Schmidt B V K J, BarnerKowollik C, Piňol M, Oriol L. Macromol., 2014, 47: 3693-3700
8 Zhang YQ, Ortega J, Baumeister U, Folcia C L, SanzEnguita G, Walker C, RodriguezConde S, Etxebarria J, O′Callaghan M J, More K. J. Am. Chem. Soc., 2012, 134(39): 16298-16306
9 Bobrovsky A, Shibaev V, Cigl M, Hamplov V, Hampl F, Elyashevitch G. J. Mater. Chem. C, 2014, 2: 4482-4489
10 Merino E, Ribagorda M. Beilstein J. Org. Chem., 2012, 8: 1071-1090
11 Sun X M, Wang W, Qiu L B, Guo W H, Yu Y L, Peng H S. Angew. Chem. Int. Ed., 2012, 51: 8520-8524
12 Gorostiza P, Isacoff E Y. Science, 2008, 322: 395-399
13 Wyart C, Del Bene F, Warp E, Scott E K, Trauner D, Baier H, Isacoff E Y. Nature, 2009, 461: 407-410
14 Hoppmann C, Seedorff S, Richter A, Fabian H, Schmieder P. Angew. Chem. Int. Ed., 2009, 48: 6636-6639
15 Beharry A A, Woolley G A. Chem. Soc. Rev., 2011, 40: 4422-4437
16 RodríguezRedondo L J, SastreSantos , FernándezLázaro F, Soares D, Azzellini G C, Elliott B, Echegoyen L. Chem. Commun., 2006, 42: 1265-1268
17 Siewertsen R, Neumann H, BuchheimStehn B, Herges R, Renth F, Temps F. J. Am. Chem. Soc., 2009, 131: 15594-15595
18 Yoshino J, Kano N, Kawashima T. Chem. Commun., 2007, 43: 559-561
19 Beharry A A, Sadovski, O, Woolley G A. J. Am. Chem. Soc., 2011, 133: 19684-19687
20 S amanta S, Woolley G A. ChemBioChem, 2011, 12: 1712-1723
21 Samanta S, McCormick M T, Schmidt K S, Seferos S D, Woolley G A. Chem. Commun., 2013, 49: 10314-10316
22 Samanta S, Babalhavaeji A, Dong M X, Woolley G A. Angew. Chem. Int. Ed., 2013, 52: 14127-14130
23 Samanta S, Qin C G, Lough J A, Woolley G A. Angew. Chem. Int. Ed., 2012, 51: 6452-6455
24 Behrendt R, Schenk M, Musiol HJ G, Moroder L. J. Pep. Sci., 1999, 5: 519-529
25 Renner C, Kusebauch U, Loweneck M, Milbradt A G, Moroder L. J. Pep. Res., 2005, 65: 4-14
26 LI YuChuan, LI ShengHua, QI Cai, ZHANG HuiJuan, ZHU MengYu, PANG SiPing. Acta Chim. Sin., 2011, 69: 2159-2165
李玉川, 李生华, 祁 才, 张慧娟, 朱梦宇, 庞思平. 化学学报, 2011, 69: 2159-2165
27 Schrader T E, Schreier W J, Cordes T, Koller F O, Babitzki G, Denschlag R, Renner C, Lweneck M, Dong S L, Moroder L, Tavan P, Zinth W. Proc. Natl. Acad. Sci. USA. 2007, 104: 15729-15734
28 Asanuma H, Liang X, Nishioka H, Matsunaga D, Liu M, Komiyama M. Nat. Protoc., 2007, 2: 203-212
29 Guerrero L, Smart O S, Weston C J, Burns D C, Woolley G A, Allemann R K. Angew. Chem. Int. Ed., 2005, 44: 7778-7782
30 Nakayama K, Endo M, Majima T. Chem. Commun., 2004, 2386-2387
31 Dong S L, Lweneck M, Schrader T E, Schreier W J, Zinth W, Moroder L, Renner C. Chem. Eur. J., 2006, 12: 1114-1120
32 Woolley G A, Jaikaran A S I, Berezovski M, Calarco J P, Krylov S N, Smart O S, Kumita J R. Biochem., 2006, 45: 6075-6084
33 Pieroni O, Fissi A, Angelini N, Lenci F. Acc. Chem. Res., 2001, 34: 9-17
34 Renner C, Moroder L. ChemBioChem., 2006, 7: 868-878
35 Aaemissegger A, Krutler V, van Gunsteren W F, Hilvert D. J. Am. Chem. Soc., 2005, 127: 2929-2936
36 Sadovski O, Beharry A A, Zhang F, Woolley G A. Angew. Chem. Int. Ed., 2009, 48: 1484-1486
37 Beharry A A, Sadovski O, Woolley G A. J. Am. Chem. Soc., 2011, 133: 19684-19687
38 Ali M A, Woolley G A. Org. Biomol. Chem., 2013, 11: 5325-5331
39 Stawski P, Sumser M, Trauner D. Angew. Chem. Int. Ed., 2012, 51: 5748-5751
40 Tochitsky I, Banghart M R, Mourot A, Yao J Z, Gaub B, Kramer R H, Trauner D. Nat. Chem., 2012, 4: 105-111
41 Stawski P, Janovjak H, Trauner D. Bioorg. Med. Chem., 2010, 18: 7759-7772
42 Kramer R H, Fortin D L, Trauner D. Curr. Opin. Neurobiol., 2009, 19: 544-552
43 Fehrentz T, Schnberger M, Trauner D. Angew. Chem. Int. Ed., 2011, 50: 12156-12182
44 Mourot A, Kienzler M A, Banghart M R, Fehrentz T, Huber F M E, Stein M, Kramer R H, Trauner D. ACS Chem. Neurosci., 2011, 2: 536-543
45 Hilf R J C, Bertozzi C, Zimmermann I, Reiter A, Trauner D, Dutzler R. Nat. Struct. Mol. Biol., 2010, 17: 1330-1336
46 Banghart M, Borges K, Isacoff E, Trauner D, Kramer R H. Nat. Neurosci., 2004, 7: 1381-1386
47 Volgraf M, Gorostiza P, Numano R, Kramer R H, Isacoff E Y, Trauner D. Nat. Chem. Biol., 2006, 2: 47-52
48 Gorostiza P, Volgraf M, Numano R, Szobota S, Trauner D, Isacoff E Y. Proc. Natl. Acad. Sci. USA, 2007, 104: 10865-10870
49 Volgraf M, Gorostiza P, Szobota S, Helix M R, Isacoff E Y, Trauner D. J. Am. Chem. Soc., 2007, 129: 260-261
50 Bredenbeck J, Helbing J, Kumita J R, Woolley G A, Hamm P. Proc. Natl. Acad. Sci. U S A., 2005, 102: 2379-2384
51 Guerrero L, Smart O S, Woolley G.A, Allemann R K. J. Am. Chem. Soc., 2005, 127: 15624-15629
52 Beharry A A, Wong L, Tropepe V, Woolley G A. Angew. Chem. Int. Ed., 2011, 50: 1325-1327
53 Crisalli P, Kool T. Bioconjugate Chem., 2011, 22: 2345-2354
54 Didenko V V. Biotechniques, 2001, 31: 1106-1121
55 Tan L, Li Y, Drake J T, Moroz L, Wang K M, Li J, Munteanu A, Yang C Y J, Martinez K, Tan W H. Analyst, 2005, 130: 1002-1005
56 Tan W H, Wang K M, Drake J T. Curr. Opin. Chem. Biol., 2004, 8: 547-553
57 Johnston P R A, Caruso F. J. Am. Chem. Soc., 2005, 127: 10014-10017
58 Liu J W, Lu Y. Anal. Chem., 2003, 75: 6666-6672
59 Medintz I L, Goldman E R, Lassman M E, Mauro J M. Bioconj. Chem., 2003, 14: 909-912
60 Hunter C A, Togrul M, Tomas S. Chem. Commun., 2004, 108-109
61 Goodman A, Breinlinger E, Ober M, Rotello V M. J. Am. Chem. Soc., 2001, 123: 6213-6214
62 Gomy C, Schmitzer R A. Org. Lett. 2007, 9(2): 3865-3868
63 Pearson D, Abell A D. Chem. Eur. J., 2010, 16: 6983-6992
64 Stawski P, Sumser M, Trauner D. Angew. Chem. Int. Ed., 2012, 51: 5748-5751
65 Tsuwi J, Berger R, Labat G, Couderc G, Behrnd N R, Ottiger P, Cucinotta F, Schurmann K, Bertoni M, Viani L, Cornil J, ProdiSchwab A, de Cola L, Wubbenhorst M, Hulliger J. J. Phys. Chem. A, 2010, 114(26): 6956-6963
66 Kim H S,Cao T, Pham T, Yoon K B. Chem. Commun., 2012, 48: 4659-4673
67 Yu G C, Han C Y, Zhang Z B, Chen J Z, Yan X Z, Zheng B, Liu S Y, Huang F H. J. Am. Chem. Soc., 2012, 134: 8711-8717
68 Dsouza N R, Nau M W. J. Org. Chem., 2008, 73: 5305-5310
69 Bakirci H, Koner L A, Nau M W. J. Org. Chem., 2005, 70: 9960-9966
70 Liu Y, Wang H, Zhang H Y, Liang P. Chem. Commun., 2004, 2266-2267
71 Zaman M B, Udachin K, Akhtaruzzaman M, Yamashita Y, Ripmeester A J. Chem. Commun., 2002, 2322-2323
72 Tomatsu I, Hashidzume A, Harada A. J. Am. Chem. Soc., 2006, 128: 2226-2227
73 Ferris D P, Zhao YL, Khashab N M, Khatib H A, Stoddart J F, Zink J I. J. Am. Chem. Soc., 2009, 131: 1686-1688
74 Liu Y, Yang ZX, Chen Y. J. Org. Chem., 2008, 73: 5298-5304
75 Harada A. Acc. Chem. Res., 2001, 34: 456-464
76 Wang D S, Zhang X X, Nie S Q, Zhao W F, Lu Y, Sun S D, Zhao C S. Langmuir, 2012, 28: 13284-13293
77 Zhang Y R, Ren L, Tu Q, Wang X Q, Liu R, Li L, Wang J C, Liu W M, Xu J, Wang J Y. Anal. Chem., 2011, 83: 9651-9659
78 Banerjee A I, Yu L T, Matsui H. J. Am. Chem. Soc., 2003, 125: 9542-9543
79 Wan P B, Chen Y Y, Xing Y B, Chi L F, Zhang X. Langmuir, 2010, 26: 12515-12517
80 Gong Y H, Li C, Yang J, Wang H Y, Zhuo R X, Zhang X Z. Macromol., 2011, 44: 7499-7502
81 Frasconi M, Mazzei F. Langmuir, 2012, 28: 3322-3331
82 Tamesue S, Takashima Y, Yamaguchi H, Shinkai S, Harada A. Angew. Chem., 2010, 122: 7623-7626
83 Liu J H, Chen G S, Guo M Y, Jiang M. Macromol. 2010, 43: 8086-8093
84 Liao X J, Chen G S, Liu X X, Chen W X, Chen F E, Jiang M. Angew. Chem. Int. Ed., 2010, 49: 4409-4413
85 Liao X J, Chen G S, Jiang M. Langmuir, 2011, 27: 12650-12656
86 Zhang H J, Xin Y, Yan Q, Zhou L L, Peng L, Yuan J Y. Macromol. Rapid Commun. 2012, 33: 1952-1957
87 Liu R, Zhang Y, Feng P Y. J. Am. Chem. Soc., 2009, 131: 15128-15129
88 Wang D S, Zhang X X, Nie S Q, Zhao W F, Lu Y, Sun S D, Zhao C S. Langmuir 2012, 28: 13284-13293
89 Jin H B, Zheng Y L, Liu Y, Cheng H X, Zhou Y F, Yan D Y. Angew. Chem. Int. Ed., 2011, 50: 10352-10356
90 Wu M, Cao Y P, Zhang X Z, Zhang Y F, Chen Y, He L, Qian Z Y. Chem. Commun., 2012, 48: 9846-9848
91 Woolley, G A. Nat. Chem., 2012, 4: 75-77
92 Woolley, G A. Nat. Nanotechnol., 2013, 8: 892-893
93 QIN ChuanGuang,ZHANG Yuan, LI Lin, SHANG XiaoYa, NIU WeiNing, XU ChuanLan. Sci. Sin. Chim., 2013, 43: 1322-1335
钦传光, 张 媛, 李 琳, 尚晓娅, 牛卫宁, 徐春兰. 中国科学: 化学, 2013, 43: 1322-1335
94 REN Jin, QIN ChuanGuang, XU ChuanLan, WANG QiuYu, ZUO XiaoJia. Act. Pharmceut. Sin., 2010, 45: 17-25
任 锦, 钦传光, 徐春兰, 王秋雨, 左晓佳. 药学学报, 2010, 45: 17-25
95 Zhang F, Timm A K, Arndt M K, Woolley G A. Angew. Chem. Int. Ed., 2010, 49: 3943-3946
96 Szobota S, Isacoff Y E. Annu. Rev. Biophys. Biomol. Struct., 2010, 39: 329-348
97 Banghart R M, Mourot A, Fortin L D, Yao J Z, Kramer R H, Trauner D. Angew. Chem. Int. Ed., 2009, 48: 9097-9101
98 Lim R K, Lin Q. Chem. Commun., 2010, 46: 1589-1600
99 Boulegue C, Loweneck M, Renner C, Moroder L. ChemBioChem, 2007, 8: 591-594
100 Bose M, Groff D, Xie J, Brustad E, Schultz P G. J. Am. Chem. Soc., 2006, 128: 388-389
101 Zhang Y, Erdmann F, Fischer G. Nat. Chem. Biol., 2009, 5: 724-726
102 Sadovski O, Beharry A A, Zhang F, Woolley G A. Angew. Chem. Int. Ed., 2009, 48: 1484-1486
103 Beharry A A, Sadovski O, Woolley G A. Org. Biomol. Chem., 2008, 6: 4323-4332
Abstract Azobenzene derivatives have received considerable experimental and theoretical attention, along with more and more the investigation and the knowledge on the characteristics of their photoinduced isomerization. Except for their traditionally wellknown uses as dyes or colorants in many industries, in recent years, azobenzene derivatives have been widely used as photoresponsive functional devices utilized not only as smart polymers, liquid crystals, molecular switches, and machines, which have been reviewed in several reports. Simultaneously, they also have been rapidly permeated and applied to the field of chemical and biological analysis as photoswitchable molecular probes and sensory chips, playing an increasingly significant role. So, it is worth emphasizing to review and summarize the last developments in the later topics as title of this paper. More than 100 relevant literatures are cited here and the prospects are pointed out.
Keywords Azobenzene derivative; Molecular probe; Sensory chip; Bioanalysis and imaging; Photoswitching agent; Molecular recognition; Review
(Received 8 October 2014; accepted 22 December 2014)
This work was supported by the National Natural Science Foundation of China (Nos. 20672086, 20802057) and the Natural Science Basic Research Plan in Shaanxi Province of China (No. 2012JZ2002)
