白蕾 霍淑慧 韩振刚 陈晶
摘 要:合成了4个双功能方酰胺探针分子,其中1和3为新化合物,2和4为已知化合物,并通过核磁共振(1H NMR、13C NMR)和高分辨质谱(HRMS)确认其结构。采用荧光光谱法,系统研究了这4个手性探针分子对Boc苯丙氨酸、苯丙氨酸、苯甘氨酸、脯氨酸、缬氨酸、酒石酸、扁桃酸和联二萘酚的荧光手性识别效果。通过组合筛选发现,探针分子4对缬氨酸有较好的识别效果。进一步的研究结果表明,探针分子4与缬氨酸的摩尔比为1∶2时,加入L缬氨酸后, 荧光光谱峰大幅蓝移,且荧光强度大幅减弱; 加入D缬氨酸后, 荧光光谱没有变化,荧光强度比值(ID/IL)达到1.35。据此提出了探针分子4的叔胺基团和方酰胺基团分别通过静电作用和氢键作用各结合一分子L缬氨酸的双手性中心识别机理。
关键词:手性识别; 荧光探针; 方酰胺; 双功能; 非衍生氨基酸1 引 言
天然的手性物质都以单一异构体的形式存在,许多生物大分子(如核酸、蛋白质、氨基酸、多糖等)都是手性的,许多关键的生物过程与手性物质的特定相互作用有关。药物分子多为手性化合物,其对映异构体常表现出不同的生理和药理活性。在非手性环境下, 对映异构体理化性质完全一致,所以识别、检测和分离手性化合物的对映异构体非常具有挑战性。相比传统的高效液相色谱、气相色谱和电泳,荧光光谱检测法具有操作简单、成本低、快速且可以高通量识别和检测手性化合物的优势[1~8]。在荧光手性探针分子中,有机小分子类探针由于易于合成和修饰、结构多变,成为手性识别领域的主要研究内容[9~18]。方酰胺是具有较强双氢键给体的四元环状化合物,其两个侧链易于修饰,已被广泛应用在不对称催化领域[19~25]。本课题组期望将手性方酰胺发展为新型的有机小分子手性荧光探针,并且报道了第一例方酰胺探针在荧光手性识别中的应用[26]。
在荧光手性识别客体中,非衍生氨基酸由于在绝大多数有机体系中难溶,其手性识别存在巨大挑战。近期,美国弗吉尼亚大学蒲林研究团队报道了在醋酸锌存在下,手性醛探针分子可与非衍生氨基酸的胺基缩合生成亚胺,由此实现了对13種天然氨基酸的高效识别[9]。本研究在方酰胺探针分子中引入碱性的叔胺基团(图1,Type 1),使其能够与氨基酸的羧基发生酸碱中和反应,由此实现对非衍生氨基酸的手性识别。另外,在探针分子中同时引入两个方酰胺片段,可构建一类具有4个氢键作用位点的口袋式探针分子(图1,Type 2)。基于此,本研究合成了两个双方酰胺探针分子(化合物1和2)和两个方酰胺叔胺探针分子(化合物3和4),其中1和3为新化合物,并研究了这4个双功能方酰胺探针分子对氨基酸及其衍生物、羟基酸和轴手性化合物联二萘酚的荧光手性识别效果。

2.1 仪器与试剂
Bruker500型核磁共振波谱仪(德国Bruker公司); 日立F7000荧光光谱仪(日本日立公司);? QTOF 6520B高分辨质谱仪(美国安捷伦公司)。
3,5二三氟甲基苯胺、(1R,2R)1,2环己二胺、(1R,2R)1,2二苯基乙二胺(分析纯,中国安耐吉公司); 方酸二甲酯、二碳酸二叔丁酯(分析纯,北京中胜华腾公司); 苯丙氨酸、Boc苯丙氨酸、苯甘氨酸、脯氨酸、缬氨酸、酒石酸、扁桃酸和联二萘酚(分析纯,北京沃尔吉明公司)。
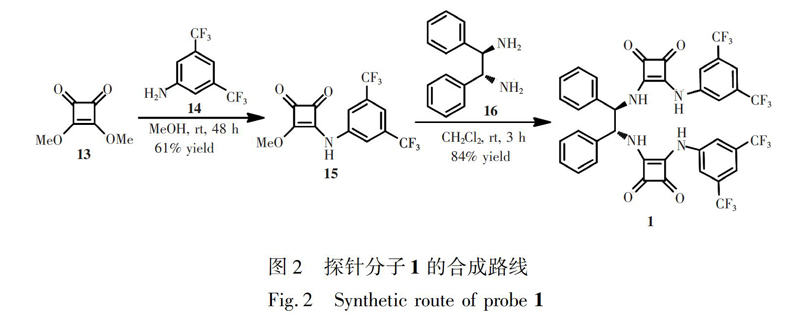
2.2 探针分子1的合成
探针分子1的合成路线如图2所示:在100 mL圆底烧瓶中加入3,5二三氟甲基苯胺14 (10 mmol, 2.29 g) 和40 mL甲醇,搅拌使其完全溶解后,再加入方酸二甲酯13 (10 mmol, 1.42 g)。在室温下继续搅拌48 h,过滤,所得的滤渣为中间体15。粗产品经甲醇二氯甲烷重结晶,得2.27 g纯品产物,产率61%。
在50 mL 圆底烧瓶中加入中间体15(2.0 mmol,0.68 g) 和10 mL二氯甲烷,搅拌使其完全溶解,再加入(1R,2R)1,2二苯基乙二胺16 (1.0 mmol,0.21 g),室温搅拌3 h,过滤,用二氯甲烷多次洗涤滤渣,得到0.75 g淡黄色的固体粉末1,产率84%。探针分子1的核磁数据如下: 1H NMR (600 MHz, DMSO) δ 8.11 (s, 2H), 7.68 (s, 1H), 7.55~7.19 (m, 15H), 5.42 (s, 2H), 4.53 (d, J=4.0 Hz, 2H); 13C NMR (150 MHz, DMSO) δ 185.10, 181.44, 170.53, 163.50, 143.23, 142.11, 141.19, 132.42, 132.20, 129.91, 129.44, 128.94, 128.35, 128.08, 128.03, 127.54, 126.80, 124.99, 123.18, 118.89, 118.56, 115.56, 110.49, 64.32, 60.40。探针分子1的高分辨质谱数据如下:ESIHRMS(C38H23F12N4O4计算值), m/z: 827.1511(827.1519)。探针分子1的核磁图谱见电子版文后支持信息图S1和S2。
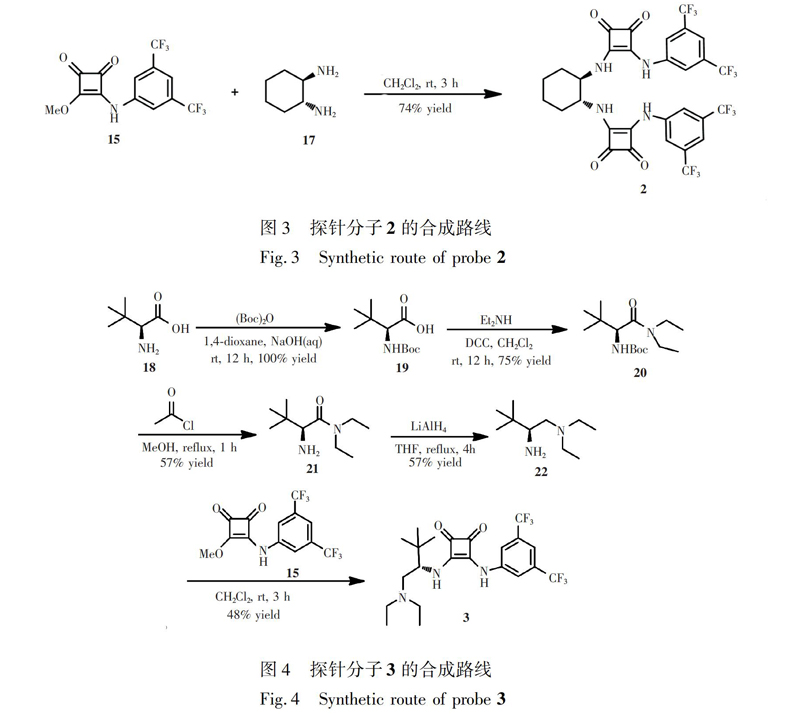
2.3 探针分子2的合成
探针分子2的合成路线如图3所示:在50 mL圆底烧瓶中加入中间体15 (2.0 mmol,0.68 g) 和干燥的二氯甲烷,搅拌使其完全溶解。再加入(1R,2R)1,2环己二胺17 (1.0 mmol,0.114 g),室温搅拌3 h,过滤,所得滤渣用二氯甲烷多次洗涤,得到0.59 g白色固体粉末2,产率74%。探针分子2的核磁氢谱与文献\[21\]一致,其数据如下:1H NMR(600 MHz, DMSO) δ 10.11~8.78(s, 2H), 7.91(s, 4H), 7.71(s, 2H), 7.61(s, 2H), 3.91(s, 2H), 2.10(s, 2H), 1.79(s, 2H), 1.44(m, 4H)。
2.4 探针分子3的合成
探针分子3的合成路线如图4所示: 参照文献\[24\]的方法合成中间体22。在250 mL圆底烧瓶中加入0.4 g NaOH和10 mL水,再加入L叔亮氨酸18 (0.1 mol),将二碳酸二叔丁酯 ((Boc)2O, 0.1 mol) 溶于10 mL 1,4二氧六环中,缓慢滴加,室温搅拌12 h,浓缩至10 mL,加入10 mL乙酸乙酯。加入3~4 mL 4 mol/L HCl,调至溶液为中性或弱酸性,搅拌3 min,分液,有机相用10 mL水洗涤,无水Na2SO4干燥,旋出溶剂,真空干燥,得到23.1 g产物BocL叔亮氨酸19,产率100%。
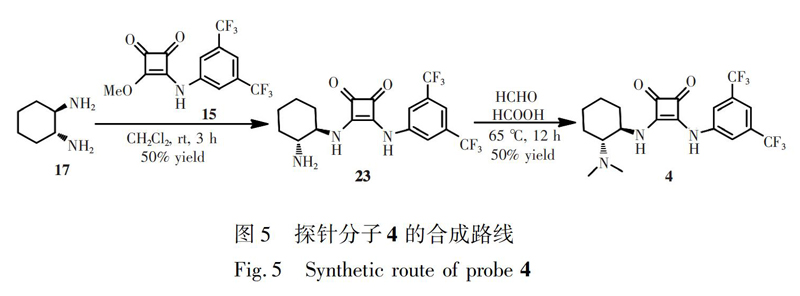
在500 mL圆底烧瓶中加入19(0.1 mol, 23.1 g)和120 mL干燥二氯甲烷,冰浴下缓慢滴加二环己基碳二亚胺 (DCC, 0.11 mol, 22.6 g) 的二氯甲烷 (100 mL) 溶液,10 min滴完。溶液變浑浊,搅拌0.5 h后, 缓慢滴加二乙胺 (0.1 mol, 7.3 g) 的二氯甲烷 (50 mL) 溶液,自然升至室温,搅拌12 h, 生成大量白色沉淀,过滤,并用二氯甲烷洗涤固体,有机相依次用2% HCl、 5% NaHCO3和饱和食盐水洗涤,无水Na2SO4干燥,蒸除溶剂,柱层析得化合物20,产率75%。
将化合物20溶于200 mL无水甲醇中,缓慢滴加20 mL乙酰氯,回流1 h后降至室温,减压除去溶剂,向剩余物中加入120 mL二氯甲烷和80 mL水。滴加2 mol/L HCl至pH=1~2,分液,收集水相。在水相中滴加K2CO3,调节至pH=11~12,加入等体积的二氯甲烷,分出有机相,再加入等体积的二氯甲烷萃取,合并有机相,无水Na2SO4干燥。蒸干溶剂,得化合物21,产率57%。

在250 mL圆底烧瓶中加入化合物21 (5.7 g) 和100 mL四氢呋喃,在冰浴下分批少量加入2.9 g四氢锂铝。缓慢升温,回流4 h。冷却至室温,冰浴下滴加饱和Na2SO4 (40 mL) 淬灭反应,抽滤除去生成的固体沉淀,乙酸乙酯萃取滤液,有机相用无水Na2SO4干燥,除去溶剂后减压蒸馏(75~78℃/0.01 kPa)得化合物22,产率57%。
在50 mL圆底烧瓶中加入中间体15 (0.1 mmol, 0.34 g),再加入20 mL二氯甲烷使其完全溶解,然后加入化合物22 (0.1 mmol, 0.2 g),室温下搅拌3 h,过滤,用二氯甲烷多次洗涤滤渣,所得滤渣即探针分子3,产率48%。探针分子3的核磁数据如下:1H NMR (600 MHz, DMSO) δ 10.10 (s, 1H), 8.42 (s, 1H), 8.11 (s, 1H), 7.63 (d, J=13.8 Hz, 2H), 4.04 (t, J=9.1 Hz, 1H), 2.63~2.44 (m, 3H), 2.41–2.30 (m, 1H), 2.22 (s, 2H), 1.37 (m, 6H), 0.95 (s, 9H); 13C NMR (150 MHz, DMSO) δ 185.51, 181.03, 171.62, 162.45, 142.15, 132.42, 132.20, 124.99, 123.18, 118.84, 115.35, 61.40, 59.76, 55.11, 34.67, 34.49, 27.11, 26.92, 26.63, 24.81。探针分子3的高分辨质谱数据如下:ESIHRMS (C22H28F6N3O2计算值), m/z 480.2080(480.2080)。探针分子3的核磁图谱见电子版文后支持信息图S3和S4。

2.5 探针分子4的合成
探针分子4的合成路线如图5[25]所示:在100 mL圆底烧瓶中加入(1R.2R)1,2环己二胺17 (1.3 mmol, 0.41 g)、干燥的二氯甲烷10 mL,然后加入中间体15 (1.3 mmol,0.14 g),室温下搅拌3 h。过滤,滤渣用乙酸乙酯/石油醚重结晶,得到白色固体0.28 g,即化合物23,产率50%。
在100 mL圆底烧瓶中加入化合物23 (1.74 mol,0.91 g),再加入甲醛(2.5 mL, 37%)和甲酸 (2.5 mL, 98%),缓慢升温至65℃。12 h后停止加热,冷却至室温,以10% NaOH 溶液调节至pH 10,过滤,滤渣用甲醇/二氯甲烷重结晶,得白色固体0.45 g,即探针分子4,产率50%。探针分子4的核磁氢谱与文献\[25\]一致,其数据如下:1H NMR (600 MHz, DMSO) δ 10.36 (s, 1H), 8.07 (s, 2H), 7.85 (s, 1H), 7.65(s, 1H), 3.78~3.93 (m, 1H), 2.36~2.46 (m, 1H), 2.19 (s, 6H), 2.04~2.14 (m, 1H), 1.80~1.90 (m, 1H), 1.71~1.79 (m, 1H), 1.63~1.70 (m, 1H)和1.13~1.41(m, 4H)。

2.6 探针分子1~4的紫外光谱
为了选择适于探针分子的荧光激发波长,首先测试4种化合物的紫外光谱,选择最大紫外吸收峰作为荧光的激发波长。以二甲亚砜(DMSO)为溶剂,将探针分子1~4配制成1.0×10-5 mol/L的溶液,测定探针分子的紫外吸收光谱(见电子版文后支持信息图S5)。探针分子1~4的紫外光谱最大吸收峰波长分别为342、341、352和346 nm。以此作为这4个探针分子的荧光激发波长。
2.7 溶液配制
以测试探针分子1对苯丙氨酸6的手性识别特性为例,探针分子1与苯丙氨酸6的摩尔比为1∶1。以DMSO为溶剂,将探针化合物1、L苯丙氨酸(6L)、D苯丙氨酸(6D)配制成浓度为1.0×10-5 mol/L溶液。将探针分子1的溶液和L苯丙氨酸(6L)的溶液等体积混合得混合液A,将探针分子1的溶液和D苯丙氨酸(6D)的溶液等体积混合得混合液B,静止1 h后,在激发波长342 nm的条件下分别观察探针分子1的溶液、混合液A、混合液B的荧光强度,观察探针分子1中分别加入L苯丙氨酸和D苯丙氨酸的荧光谱图变化,判断探针分子1对苯丙氨酸两种对映异构体的识别效果。3 结果與讨论
3.1 探针分子1~4的荧光手性识别
将化合物1~4作为探针分子,分别对氨基酸及其衍生物(Boc苯丙氨酸5、苯丙氨酸6、苯甘氨酸7、脯氨酸8、缬氨酸9)、羟基酸(酒石酸10、扁桃酸11)和轴手性化合物联二萘酚12进行荧光手性识别研究(图6)。
实验结果表明,探针分子1~4在分别加入LBoc苯丙氨酸和DBoc苯丙氨酸后荧光强度没有明显变化(见电子版文后支持信息图S6)。探针分子1、2、4在加入L苯丙氨酸和D苯丙氨酸后荧光强度也没有明显变化,探针分子3在加入L苯丙氨酸后荧光强度略微增强,但加入D苯丙氨酸后荧光强度没有变化,二者的荧光强度比IL/ID=1.13(见电子版文后支持信息图S7)。探针分子1~4对苯甘氨酸和脯氨酸的两个对映异构体均没有明显的识别效果(见电子版文后支持信息图S8和图S9)。探针分子1和2在加入L缬氨酸和D缬氨酸后荧光强度没有明显变化; 探针分子3在分别加入L缬氨酸和D缬氨酸后,荧光峰都发生了蓝移,但二者相比没有明显变化; 探针分子4在加入L缬氨酸后荧光强度有较大幅度的增强,但加入D缬氨酸后荧光强度没有变化,二者的荧光强度比IL/ID=1.20(图7)。
探针分子1~4对酒石酸和扁桃酸的两个对映异构体均无明显识别效果(见电子版文后支持信息图S10和S11)。探针分子1~4在分别加入R联二萘酚和S联二萘酚(见电子版文后支持信息图S12)后,荧光光谱的峰都发生了大幅蓝移,但二者强度相比没有变化,可能是由于联二萘酚的荧光强度比探针分子更大,掩盖了探针分子的峰。
通过测定探针分子1~4与手性化合物5~12对映异构体的荧光响应谱图,计算两种异构体荧光谱图强度的比值(IR/IS),结果见表1。通过对比发现探针分子对缬氨酸9的两个对映异构体有较好的识别效果,IL/ID=1.20。另外,探针分子3对苯丙氨酸6也有比较明显的识别效果。
3.2 探针分子4与不同摩尔比的缬氨酸荧光手性识别
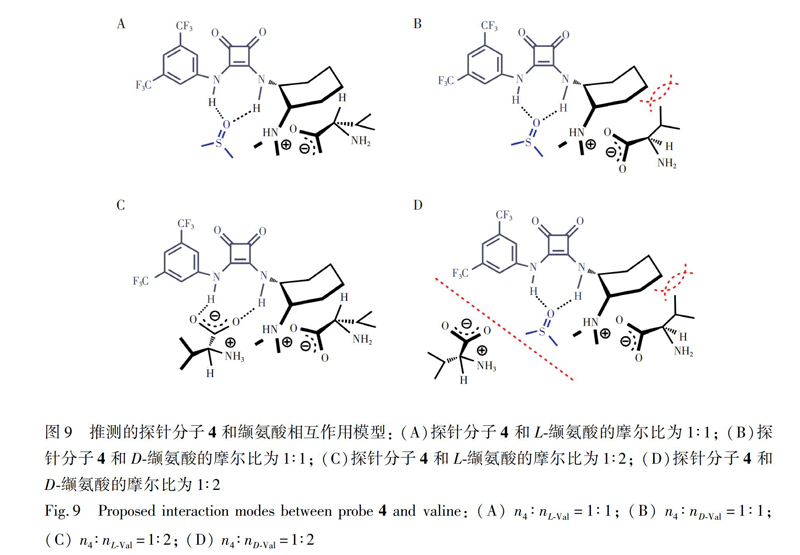
分别测定了探针分子4与缬氨酸的摩尔比为 1∶0.5、 1∶1、1∶2、1∶3时的识别效果(图8)。结果表明, 探针分子4与缬氨酸的摩尔比由1∶1变为1∶0.5时,加入L缬氨酸和D缬氨酸荧光强度都有所增强,但IL/ID=1.05。探针分子4与缬氨酸的摩尔比由1∶1变为1∶2时,加入L缬氨酸后,荧光光谱峰大幅度蓝移,最高峰由528 nm移到463 nm,而且荧光强度也大幅减弱; 加入D缬氨酸后,荧光光谱几乎没有变化,IL/ID=1.35。探针分子4与缬氨酸的摩尔比由原来的1∶1变为1∶3时,加入L缬氨酸和D缬氨酸荧光强度都大幅减弱,但二者几乎没有区别,IL/ID=1.01。由此可知,探针分子4与缬氨酸的摩尔比1∶2时,识别效果最好,加入L缬氨酸后,荧光光谱峰大幅蓝移,而且荧光强度大幅减弱,加入D缬氨酸后荧光光谱几乎没有变化。
3.3 探针分子4识别缬氨酸异构体的机理推测
上述实验表明,探针分子4对缬氨酸的2个对映异构体有较好的手性识别效果,在二者摩尔比为1∶1时,IL/ID=1.2; 摩尔比为1∶2时,IL/ID=1.35。据此推测,当探针分子4与L缬氨酸按摩尔比1∶1混合后,L缬氨酸的羧基会与探针分子4的叔胺基团发生酸碱中和反应,将羧基质子转移到叔胺上,在溶液中以阴阳离子对的形式存在,方酰胺的两个氢键位点则被溶剂DMSO占据(图9A)。当探针分子4与D缬氨酸按摩尔比1∶1混合后,D缬氨酸仍然会与探针分子4的叔胺基团形成阴阳离子对,但D缬氨酸的异丙基与探针分子4的环己烷在同侧,空间位阻较大,致使二者结合不够紧密(图9B)。而L叔亮氨酸则是氢原子与环己烷在同侧,空间位阻小,二者结合比较紧密。因此,前者荧光增强,后者几乎不变。当探针分子4与L缬氨酸按摩尔比1∶2混合后,一分子L叔亮氨酸与探针分子中的叔胺基团形成盐,另一分子L叔亮氨酸的羧基与探针分子中方酰胺的两个NH键形成双氢键,致使探针分子4的荧光减弱,且发生蓝移(图9C)。将探针分子4与D缬氨酸按摩尔比1∶2混合后,一分子D叔亮氨酸与探针分子中的叔胺基团形成盐,另一分子D叔亮氨酸由于空间构型与方酰胺的立体空腔不匹配,无法进入,空腔仍由溶剂DMSO占据,所以荧光光谱没有明显变化(图9D)。探针分子4有两个手性中心,当探针分子4与缬氨酸摩尔比为1∶1时,是叔胺单手性中心识别; 而探针分子4与缬氨酸摩尔比为1∶2时,是叔胺方酰胺双手性中心识别。
4 结 论
合成了两个双方酰胺探针分子和两个方酰胺叔胺探针分子,其中1和3为新化合物,2和4为已知化合物。系统研究了这4个手性探针分子对氨基酸及其衍生物、羟基酸、联二萘酚的荧光手性识别效果。通过组合筛选,发现探针分子4对缬氨酸有较好的识别效果,探针分子4与缬氨酸的摩尔比为1∶2时,加入L缬氨酸后荧光光谱峰大幅蓝移,而且荧光强度大幅减弱; 加入D缬氨酸后荧光光谱没有变化,ID/IL=1.35。据此提出了探针分子4的叔胺基团通过静电作用和方酰胺基团通过氢键作用各结合一分子L缬氨酸的双手性中心识别机理。本研究为非衍生氨基酸的荧光手性识别提供了叔胺方酰胺双功能探针的新策略,也为有机小分子探针的设计提供了多功能型复杂探针的新思路。References
1 Busschaert N, Caltagirone C, Rossom W V, Gale P A. Chem. Rev., 2015, 115(15): 8038-8155
2 Saleem M, Lee K H. RSC Adv.,? 2015,? 5: 72150-72287
3 MartínezMez R, Sancenón F. Chem. Rev.,? 2003,? 103(11): 4419-4476
4 Quang D T, Kim J S. Chem. Rev.,? 2010,? 110(10): 6280-6301
5 Gunnlaugsson T, Glynn M, Tocci G M, Kruger P E, Pfeffer F M. Coord. Chem. Rev.,? 2006,? 250(2324): 3094-3117
6 LIU DaLiang, ZHANG Chun, WEI SiPing, WANG Ping, YOU Qiang, WANG Li. Journal of Analytical Science, 2018, 34(5): 621-626
刘大亮, 张 春, 韦思平, 王 平, 尤 强, 王 力. 分析科学学报, 2018, 34(5): 621-626
7 Wu J S, Liu W M, Ge J C, Zhang H Y, Wang P F. Chem. Soc. Rev.,? 2011,? 40: 3483-3495
8 Chen X Q, Pradhan T, Wang F, Kim J S, Yoon J Y. Chem. Rev.,? 2012,? 112(3): 1910-1956
9 Zhu Y Y, Wu X D, Gu S X, Pu L. J. Am. Chem. Soc.,? 2019,? 141 (1): 175-181
10 Wen K L, Yu S S, Zeng H, Chen L M, Xiao M, Yu X Q, Pu L. J. Am. Chem. Soc.,? 2015,? 137 (13): 4517-4524
11 Lin J, Hu Q S, Xu M H, Pu L. J. Am. Chem. Soc.,? 2002,? 124(10): 2088-2089
12 Xu M H, Lin J, Hu Q S, Pu L. J. Am. Chem. Soc.,? 2002,? 124(47): 14239-14246
13 Li Z B, Lin J, Pu L. Angew. Chem. Int. Edit.,? 2005,? 44(4): 1690-1693
14 Liu H L, Peng Q, Wu Y D, Chen D, Hou X L, Sabat M, Pu L. Angew. Chem. Int. Edit.,? 2010,? 49(3): 602-606
15 Bencini A, Coluccini C, Garau A, Giorgi C, Lippolis V, Messori L, Pasini D, Puccioni S. Chem. Commun.,? 2012,? 48: 10428-10430
16 Mei X F, Wolf C. J. Am. Chem. Soc.,? 2004,? 126(45): 14736-14737
17 Zhao J Z, Fyles T M, James T D. Angew. Chem. Int. Edit.,? 2004,? 43(26): 3461-3464
18 Lu Q S, Dong L, Zhang J. Li J, Jiang L, Huang Y. Qin S, Hu C W, Yu X Q. Org. Lett.,? 2009,? 11(3): 669-672
19 Malerich J P, Hagihara K, Rawal V H. J. Am. Chem. Soc.,? 2008,? 130(44): 14416-14417
20 Albrecht , Dickmeiss G, Acosta F C, RodríguezEscrich C, Davis R L, Jrgensen K A. J. Am. Chem. Soc.,? 2012,? 134(5): 2543-2546
21 Ni X, Li X, Wang Z, Cheng J P. Org. Lett.,? 2014,? 16(6): 1786-1789
22 Zhao B L, Li J H, Du D M. Chem. Rev.,? 2017,? 17(10): 994-1018
23 Ramalingam V, Domaradzki M E, Jang S, Muthyala R S. Org. Lett.,? 2008,? 10(15): 3315-3318
24 Li Y, Yang G H, He C Q, Li X, Houk K N. Org. Lett.,? 2017,? 19(16): 4191-4194
25 Konishi H, Lam T Y, Malerich J P, Rawal V H. Org. Lett.,? 2010,? 12(9): 2028-2031
26 BAI Lei, HUO ShuHui, CHEN Jing, LU XiaoQuan. Chem. J. Chin. Univ.,? 2019,? 40(1): 41-46
白 蕾, 霍淑慧, 陳 晶, 卢小泉. 高等学校化学学报, 2019,? 40(1): 41-46
- 新时代流动人员人事档案信息化建设探析
- 信息时代创新档案管理工作的相关策略
- 大数据环境下档案社会化服务探究
- 档案信息化建设存在的问题及对策分析
- 计算机技术在数字博物馆中的应用分析
- 大数据背景下军工企业档案信息化管理探究
- 企业电子档案单套制管理探究
- 医疗保障基金中心档案合并与信息化建设刍议
- 加强档案管理安全保密以及信息化档案管理的思考
- 探讨医疗设备档案信息化管理
- 人事档案信息化管理与服务工作之探析
- 利用大数据创新档案管理模式和提升服务能力
- 疾控中心档案管理的信息化和科学化建设探讨
- 商业银行档案管理分析
- 卫生监督档案管理中的问题与对策
- 浅谈临床医学专业认证背景下医院教学档案的作用及建设
- 博物馆数字化时代档案管理的创新与优化模式思考探究
- 浅谈涉密文件的管理现状及完善措施
- 高校干部人事档案管理系统建设探究
- 基于人才流动的人事档案管理问题探究
- 机关档案收集工作中存在的问题与对策
- 论数字化时代档案的安全管理
- 浅析档案管理在企事业单位效能化发展中的作用
- 刍议事业单位办公室档案日常管理与利用
- 民政局档案管理的重要性及有效措施
- reticketing
- retickets
- retie
- retied
- retieing
- reties
- retighten
- retightened
- retightening
- retightens
- retile
- retiled
- retiles
- retiling
- retimber
- retimed
- retimes
- retiming
- re-tin
- retin
- retina
- retinae
- retinal
- retinas
- retinge
- 天不生无禄之人,地不长无名之草
- 天不生无禄之人, 地不长无名之草。
- 天不生无禄之人,地不长无根之草
- 天不盖,地不载
- 天不着边,地不着界
- 天不管地不管
- 天不能总晴,人不能常壮
- 天不能成全好事
- 天不能移,地不能动
- 天不言而人推高,地不言而人推厚,四时不言而百姓期焉
- 天不言而自高,地不言而自卑
- 天不言而自高, 地不言而自卑。
- 天不言自高,地不言自厚
- 天不言,地不语
- 天不转地转
- 天不长眼
- 天不颇覆,地不偏载
- 天与不取,反受其咎
- 天与不取,反受其咎;时至不行,反受其殃
- 天与人归
- 天与地平线交会处
- 天业
- 天丝
- 天丧斯文
- 天中