李宗瑾 宋长新


摘要:肺动脉高压(pulmonary arterialhypertension,PAH)是一种严重的心血管疾病,发病机制复杂,分子机制尚未完全阐明,临床死亡率仅次于肿瘤与心肌梗死,严重威胁人类的生命健康。因此,需要更多的方法来探究PAH的分子机制,挖掘影响其发生的关键因子。该文将PAH疾病的分子研究与加权基因共表达网络(WGCNA)相结合,通过构建PAH加权基因共表达网络,探讨其潜在生物标志物,筛选可能与PAH显著相关的关键基因,为PAH的预防及早期治疗提供理论与实践基础。
关键词:PAN;WGCNA;关键基因;生物标志物
中图分类号:TP391文献标识码:A
文章编号:1009-3044(2020)15-0008-02
1背景
PAH是一种病因不明、受遗传与环境等多因素共同影响的综合征。其特征是肺动脉压力和血管阻力增加导致右心衰竭和死亡,诊断后平均生存时间为2.8年。近年来,随着对PAH发病机制的深入研究,发现遗传因素在其发病中起着重要作用。目前,仍存在治疗难度大、药物敏感性低及预后极差的问题,因此需要更多方法来发现潜在的关键(枢纽)基因,进而揭示其分子机制,进而降低其死亡率。WGCNA是一种系统生物学方法,是以基因间表达相似性为基础来实现基因网络构建的算法,常用来探索基因和表型之间复杂关系,不仅可以筛选具有价值的生物标志物或靶位点的鉴定,还具有揭示生物学规律、疾病关键驱动因子和药物作用机理的功能。在本研究中,我们利用GEO数据库中的PAH表达谱数据构建了加权基因共表达网络,筛选可能影响PAH疾病发生的关键因子,为实现PAH的早期预防与及时治疗提供理论基础。
2材料与方法
2.1数据来源及数据预处理
本文的PAH基因表达谱数据fGSEll7261)来自GEO数据库(http://www.ncbi.nlm.nih.gov/geo,),包含25个正常人类样本,和58个PAH样本,平台为GPL6244。本研究使用R编程语言(v3.6.2),Bioconductor软件包和WGCNA軟件包进行数据分析。
2.2共表达网络的构建和模块构建
我们使用R中的WGCNA软件包来构建PAH加权基因共表达网络。首先,计算所有基因对的相关性构建Pearson相关矩阵,如公式(1)。然后,使用幂函数nil=Allg将相关矩阵转换为邻接矩阵,其中β是一个可以强调强相关性的软阈值,软阈值p可以使网络符合标准的无尺度网络,从而实现无标度拓扑。第三,将邻接矩阵转换为拓扑重叠矩阵(TOM),如公式(2),并计算基因间相异度矩阵dissTOM=1-TOM,对dissTOM层次聚类得到系统聚类树,即将具有相似表达的基因分为同簇。第四,通过dynamic Tree Cut动态剪枝算法来区分共表达模块,其中min-ModuleSize为50,deepSplit为2,其他参数设置为默认值,并计算代特征向量值(ME),通过聚类合并具有高度相似的模块,便可得到基因模块。
其中,Bij表示基因i和基因j之间的邻接系数;lij表示基因i与基因i所有共同相邻基因的邻接系数的乘积之和;ki代表基因i与所有相邻节点的邻接系数综合。若值为0,表示这两个基因都是孤立的。若值是1,表示这两个基因与所有基因都是相邻的。
2.3筛选共表达网络关键模块
本文将使用两种方法来鉴别与高血压相关的模块。第一种方法,计算各模块的模块特征基因(module eigengene,ME)与疾病性状皮尔森相关系数及其P值来确定关键模块;第二种方法,通过计算基因显著性(Gene Significance,GS)和模块显著性(Module Significance,MS)来确定关键模块。GS是指基因的表达量与某一临床信息的皮尔森相关系数,MS是指模块中所有基因的GS的平均值。一般所有模块MS与GS的绝对值越大,说明与PAH疾病越相关。
2.4鉴定关键模块的枢纽基因
模块的枢纽基因是与疾病最紧密相关的基因,通常具有更大的生物学意义。Gs表示了基因在各种网络中的重要性,模块身份(Modularmembership,MM)用来衡量基因在模块内的重要性,基因之间的权重显著性p.weighted可以通过WGCNA包中的networkScrenningO函数计算得到。为了鉴定出更加可靠的关键基因,我们再次采用网络的最大集团度(MCC),如公式(3)。筛选关键模块中的枢纽基因,最后将两种方法的共同基因作为关键基因。
3结果
3.1芯片数据预处理
我们使用R软件和limma软件包对数据进行校正批处理、去除空探针及重复基因。根据映射的探针确定了每个基因的表达值,如果多个探针映射到同一基因,则最大值表达值用于表示该基因的表达水平。最后,获得了20359个基因的表达谱。对表达谱矩阵进行标准差(sD)排序,选取前5000个基因作为共表达网络构建的输人数据,进行聚类分析,发现去除离群样本GSM3290090。
3.2并加权共表达网络的构建
软阈值的选择是构建WGCNA的关键。我们对从1到20的阈值进行了网络拓扑分析,发现当B=6时,满足无标度网络条件。通过层次聚类将具有相似表达的基因分组为同一模块,同时将切割线MEDissThres设置为0.2来合并模块,鉴定了17个基因模块。
3.3确定关键基因模块
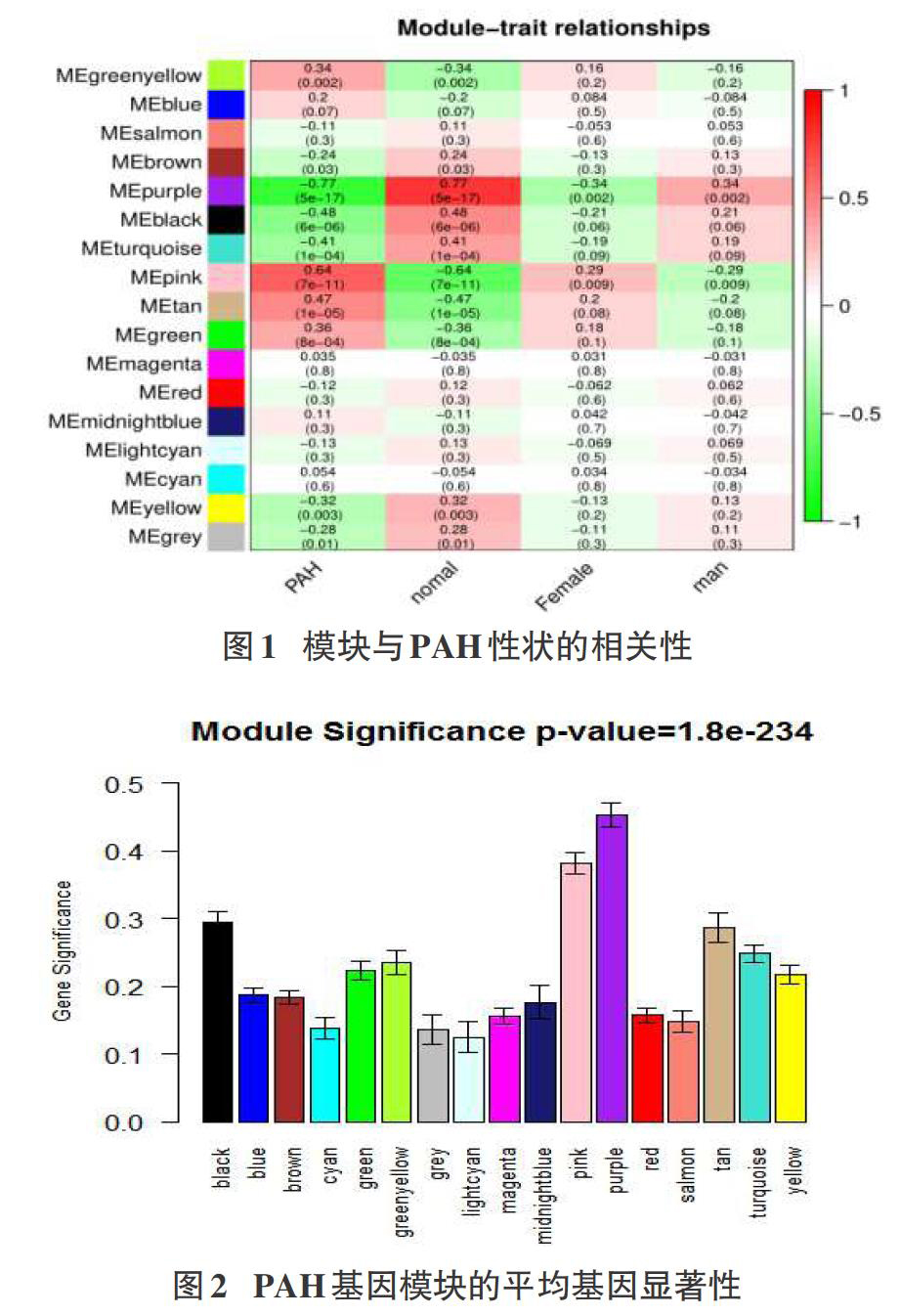
我们通过第一种方法得到从模块purple和pink与PAH相-关性系数分别为-0.77,0.64,其绝对值大于其他模块的相关系数(图1),且P值都小于0.01,因此推断这两个模块与PAH最相关,且分别与PAH在显著的负相关和正相关,即前者模块中的基因可能抑制PAH的发生,后者可能促进PAH的发生。为保证关键模块筛选的准确性,我们用另一种方法重新筛选关键模块,结果表明purple和pink模块的GS绝对值最大(图2)。用上述两种不同方法筛选结果相同,因此purple和pink模块为关键模块。
3.4与PAH发生相关的枢纽基因
在本研究中,首先以|MM|>0.8、|GS|>0.2和p.weighted<0.05为标准进行筛选,分别在purple和pink模块中筛选出了24个和21个枢纽基因。然后将关键模块基因文件导人Cyto-scape,通过MCC算法分别获得toplO枢纽基因,将两个方法获得基因取交集,最终在purple模块中确定了7个关键基因(TLR1,AQP9,SIGLEC9,LRRC25,LILRA6,LILRB3,S100A9),在pink模块中确定了5个关键基因(ANTXRl,UACA,ECM2,RBFOX2,NREP)。
4讨论
在这项研究中,我们使用WGCNA构建PAH的基因共表达网络,目的是寻找新的和关键的生物标志物。在WGCNA中,动态树切割共识别了17个基因模块,其中purple和pink模块是与PAH高度相关的两个模块。然后筛选了12个与PAH相关的潜在枢纽基因,即TLRl、AQP9、SIGLEC9、LRRC25、LILRA6、HLRB3、S100A9、ANTXRl、UACA、ECM2、RBFOX2、NREP。其中,前七个关键基因与PAH呈负相关,可能抑制PAH的发生,后五个关键基因与PAH呈正相关,可能诱发PAH的发生。
通过查询文献及基因数据库,发现有三个关键基因(TLRl、AQP9和RBFOX21已经有研究表明与PAH或高血压相关,如Koupenova M等人发现不同性别的血小板TLR转录物与不同的心血管危险因素和循环炎症水平相关;AQP9在内皮细胞,血管平滑肌细胞和心血管系统心脏中表达,参与心血管功能和相关疾病的病理过程;Zhou Y等人研究发现RBFOX2在高血压中起关键作用。有四个关键(sIGLEC9、ANTXRl、LRRC25和ECM21虽然还未有报道其与PAH相关,但其对血管具有调节作用或与TGT-β相关。其余五个关键基因(LILRA6、LILRB3、S100A9、UACA和NREPl调控参与肿瘤的免疫反应、胰岛素抵抗/2糖尿病、调节细胞凋亡、肾纤维化的新靶标等。表明我们的研究可以筛选出具有生物学意义的PAH关键基因。这些发现有助于更好地了解PAH的发病的机制,为临床决策提供预后生物标志物。
- 对县级国有投资公司战略转型的思考
- 中小企业核心竞争力的建设研究
- 中小企业品牌经营策略研究
- 企业混合所有制改革法律问题研究
- 中国农产品市场多元化发展策略研究
- 当前税收管理亟待研究和解决的几个问题
- 城市轨道交通对土地利用的作用
- 中国城市化路径与城市规模的经济学分析
- 区域经济发展视角下高校学生创新创业价值观形成策略
- 关于政府与市场关系问题的讨论——基于湖水自净现象的思考
- 浅议政府公共政策对市场的监管和推动作用
- 特色乡村旅游与农村经济发展
- 地域特色发展背景下开封市黄河文化的保护、传承和弘扬
- 基于PPP模式的体育公共服务体系构建研究
- 影响居民收入分配差距的家庭因素研究——基于家庭追踪调查数据库的考察
- 产教融合背景下高职跨境电商人才培养的实践研究——以南京工业职业技术大学为例
- 大数据时代高校人力资源管理创新研究
- 人力资源战略性管理中人力资源的定位分析
- 探析事业单位改制背景下地质工程企业人力资源管理
- 企业预算管理与绩效考核融合运用研究
- 平衡计分卡在研发企业应用——以KJ公司为例
- 高速公路养护企业人力资源管理激励机制的构建分析
- 水利水电公司人力资源绩效管理问题及对策
- 智慧城市建设的人才需求与发展分析
- 基于胜任力模型的企业人力资源招聘体系研究
- rider
- ridered
- riderless
- riders
- rides
- ride sb's/sth's coattails
- ride sth out
- ride sth ↔ out
- ride²
- ride¹
- ridge
- ridgelike
- ridger
- ridges
- ridgewise
- ridging
- ridic
- ridicle
- ridicule
- ridiculed
- ridiculer
- ridiculers
- ridicules
- ridicule²
- ridicule¹
- 分别部居
- 分别门户
- 分别附加定语
- 分别,相别
- 分到利益好处
- 分剑追财
- 分剖
- 分割
- 分割为小段
- 分割占据
- 分割土地
- 分割式
- 分割疆土
- 分割肉
- 分力
- 分加关系
- 分包
- 分化
- 分化字
- 分化瓦解
- 分区
- 分区划界
- 分厂
- 分厘
- 分厘毫丝