多接收电感耦合等离子体质谱仪测定稳定硅同位素
张安余 张经 张瑞峰 薛云

摘 要 建立了应用多接收电感耦合等离子体质谱仪(MC-ICP-MS)测定稳定硅同位素比值的仪器分析方法。在干等离子体、中分辨率条件下,硅同位素高质量端受到C、N、O、H等元素形成的多原子离子的干扰。样品气流量对Si的灵敏度稳定性有重要影响,并且样品气流量增加会导致14N16O的强度增大。中分辨率条件下,低质量数端δ29Si和δ30Si在约9 milli-amu的质量范围内保持稳定,精度优于0.04‰ (1σ)。采用“标准-样品”交叉法正校质量歧视时,为避免浓度效应对硅同位素测试的影响,要求标准和样品之间硅浓度差异低于20%。溶液酸度和Cl基体含量不会对同位素测量造成显著影响。通过优化数据采集参数,29Si/28Si和30Si/28Si的内精度(1σ)可达到8 ppm以内。标准物质长期分析结果显示,δ29Si和δ30Si的长期稳定性可达到0.06‰~0.10‰(2σ,n=20),标准物质GBW04421和GBW04422的测量值与推荐值吻合,表明本方法精确可靠。对淡水(河水、湖水)、半咸水、海水的分析结果表明,应用硅同位素可以示踪天然水体中硅的生物地球化学过程。
关键词 多接收; 硅同位素; 电感耦合等离子体质谱
1 引 言
硅(Si)是地球上丰度仅次于氧的第二大元素,约占地壳总质量的27.6%。自然界中硅循环过程(如硅酸盐风化、硅藻的光合作用)与碳循环紧密相连,在不同时间尺度上影响控制着大气CO2分压和全球气候变化[1,2]。Si具有三个稳定同位素:28Si(92.23%)、29Si(4.67%)、30Si(3.10%)[3]。测定天然水体(如河水、海水、地下水)中溶解态硅同位素组成,可以示踪硅酸盐化学风化、溶解态硅(DSi)生物矿化等硅的生物地球化学循环过程。
最初,硅同位素组成是通过将Si转化为SiF4气体并用稳定同位素质谱仪(IRMS)进行测量[4]。这种方法测量精度高,但氟化过程对实验操作安全要求高,并且所需样品量大,在分析DSi低浓度样品(如表层海水)时受到限制。近年来,多接收电感耦合等离子体质谱法(MC-ICP-MS)的迅速发展使得人们可以利用更少的样品量实现同位素的精确测量。在应用MC-ICP-MS分析硅同位素时,测量结果的精密度和准确度主要受到质谱干扰和仪器质量歧视两个因素的影响[5]。在ICP-MS中,28Si, 29Si和30Si受到来源于溶剂和大气的C、H、O、N几种元素所形成的多原子离子质谱干扰[6],特别是采用湿等离子体进样系统时,高强度的14N16O会导致30Si无法准确测量[7]。ICP-MS的仪器质量歧视主要由空间电荷效应和喷嘴效应导致[8],常用“标准-样品”交叉法(Standard-sample-bracketing,SSB)进行校正。但是,样品基体组成、待测元素浓度变化以及仪器设置上的差异均可引起质量歧视的变化。
本研究系统讨论了应用MC-ICP-MS测定硅同位素比值时多原子离子干扰、仪器参数、浓度效应、样品酸度和基体组成的影响,利用硅同位素标准物质评估了仪器分析精密度和准确度,并测定了典型水体环境中溶解态硅同位素组成。
2 实验部分
2.1 仪器与试剂
NEPTUNE多接收电感耦合等离子体质谱仪(Thermo Fisher Scientific公司);Apex-IR去溶剂装置, 100 μL/min PFA微流同心雾化器(Elemental Scientific, 美国); 硅同位素国际标准物质NBS28; 硅同位素国家标准物质GBW04421和GBW04422(北京地质矿产研究所); 99.99% Na2CO3(国药集团化学试剂有限公司);30%Supra盐酸(Merck, 德国);Milli-Q超纯水(18.2 MΩ·cm,Millipore, 美国);Dowex 50W-X8阳离子交换树脂(Sigma Aldrich公司, 美国)。
硅同位素标准溶液采用碱融法[9]进行制备:准确称取214.8 mg硅同位素标准固体样品和757.1 mg Na2CO3固体,置于铂金坩埚内混匀后950℃熔融1 h;冷却后,加入少量Milli-Q水并稍微加热使其溶解;将溶液转移至容量瓶中,用Milli-Q水润洗坩埚内壁5次,加入10 mL浓HCl,用Milli-Q水定容至100 mL,配制成1000 mg/L标准储备液;储备液转移至LDPE瓶中4℃冷藏保存。硅同位素标准溶液采用Milli-Q水为溶剂逐级稀释至500 ng/mL用于仪器测定。
2.2 样品采集与前处理
天然水体样品(河水、半咸水、海水、湖水)采集后立即用0.4 μm聚碳酸酯膜过滤,滤液置于洁净聚乙烯瓶中,盐酸酸化至pH≈2保存。
DSi>20 μmol/L的淡水样品(湖水、河水)采用阳离子交换树脂法对DSi进行纯化,半咸水、海水以及DSi<20 μmol/L的河水样品采用优化的单次氢氧化镁共沉淀(MAGIC)和阳离子交换树脂法进行富集与纯化,具体实验条件参见文献[10]。DSi浓度采用硅钼蓝比色法[11]测定。
2.3 仪器分析方法
NEPTUNE MC-ICP-MS可在静态模式下同时检测 28Si、29Si和30Si。为降低14N16O的产率,硅同位素采用干等离子体条件进行测试,样品溶液通过100 μL/min PFA雾化器和Apex-IR去溶剂装置(注:未安装膜去溶模块)引入等离子体。每次测试前用含有Si的溶液对进样系统和透镜参数进行优化,以获得最佳精密度和稳定性以及质量分辨率,具体参数设置见表1。
应用MC-ICP-MS测定硅同位素时常采用SSB法和Mg内标法校正仪器的质量歧视。由于Mg和Si的质量歧视程度不完全一致,采用Mg内标法校正质量歧视时通常需要结合SSB法[7,12]。此外,MC-ICP-MS仪器需要通过跳峰才能实现Mg和Si同位素的测定,如此会增加测试时间,减少分析通量。本研究采用SSB法校正仪器质量歧视,硅同位素组成以相对于NBS28的千分差表示,公式如下:
3 结果与讨论
3.1 质谱干扰
干等离子条件下,采用中分辨率扫描Milli-Q水空白溶液,在3个硅同位素高质量数端均存在明显的质谱干扰(图1)。丰度最高的28Si的信号稳定平台宽度约22.6 milli-amu,右侧主要干扰离子是12C16O和14N14N,二者的信号强度均低于20 mV。29Si和30Si的信号稳定平台宽度分别达到33.4和29.2 milli-amu。29Si的主要干扰离子是12C16O1H和14N14N1H,信号强度约1~2 mV,30Si的主要干扰离子为14N16O,信号强度远高于空白溶液中30Si的信号,达到20 mV。14N16O的强度呈现较大波动,可能反映了Apex-IR去除溶剂H2O时的波动。
3.2 仪器参数优化
3.2.1 样品气流量
样品气流量对待测元素灵敏度、信号稳定性和多原子离子产率有重要影响。结果显示,当样品气流量为0.95~0.97 L/min时硅同位素灵敏度和信号稳定性均达到最佳状态(图2a)。过低或者过高的样品气均会导致灵敏度和稳定性有所下降,原因在于样品气过低会造成雾化器反向压力过大,降低样品提升速率和雾化效果的稳定性,而过高的样品气会减小样品在等离子体中的停留时间,影响电离效果。14N16O强度则随着样品气流量增强而持续增加,导致30Si/14N16O的比值不断降低(图2b),这是因为增加样品进样速率时会同时引入更多的溶剂,从而增加氧化物的产率。为确保丰度最低的30Si信号强度达到80 mV左右,同时避免生成过高的14N16O,本研究将样品气流量设在0.96 L/min。
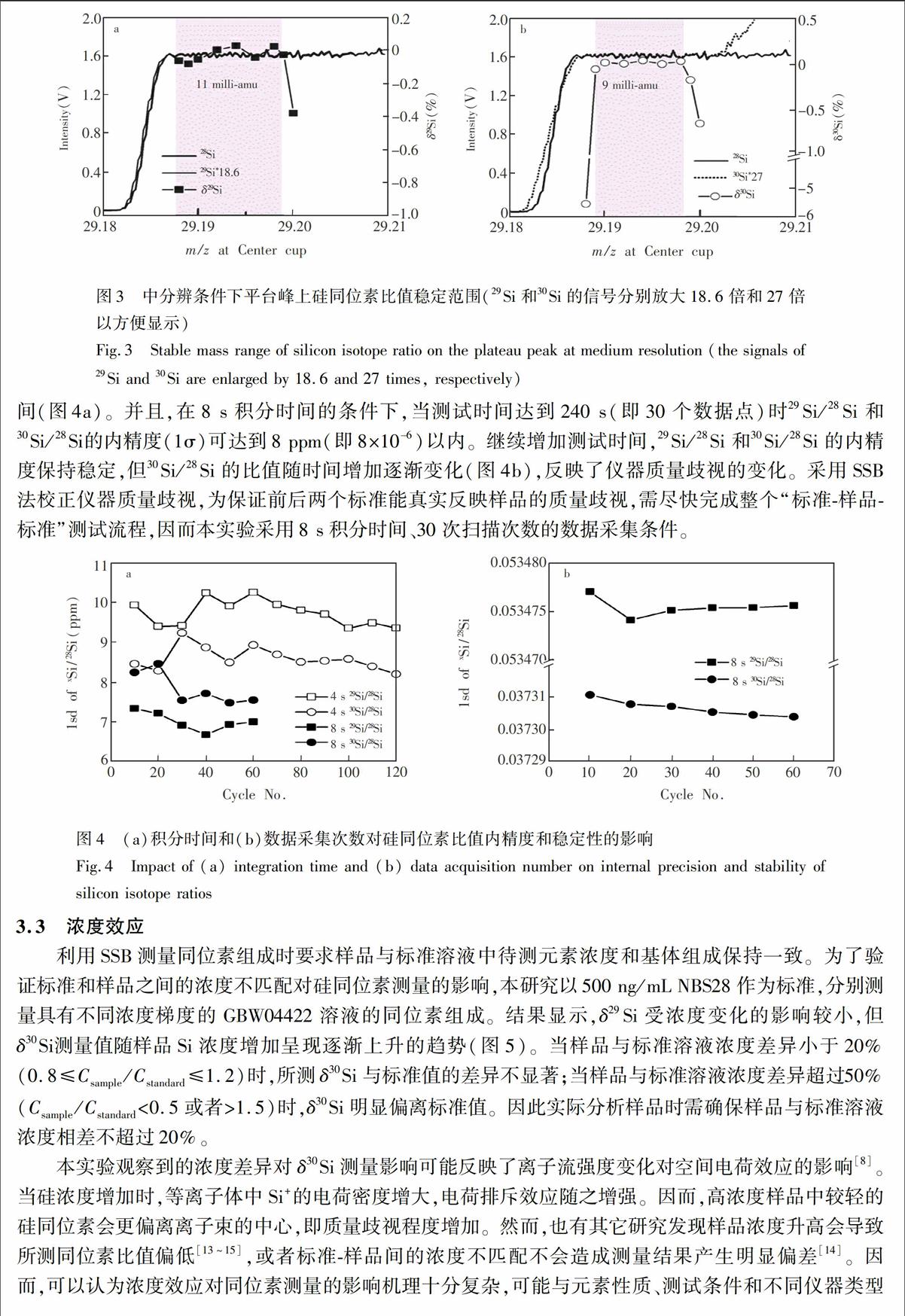
3.2.2 峰平台比值稳定性
图3展示了干等离子条件下扫描含有500 ng/mL Si溶液中硅同位素质谱图。多原子离子干扰对28Si、29Si信号强度影响不大,但14N16O会造成30Si高质量数端信号上升。为准确评估多原子离子贡献对比值的影响以及硅同位素比值稳定的质量范围,以29.192处的比值作为标准,采用SSB法测定峰平台上其它质量数处的同位素组成。结果显示,δ29Si和δ30Si分别在29.188~29.199和29.189~29.198的范围内保持稳定,同位素比值稳定的质量范围达到9 milli-amu(图3)。在比值稳定的质量范围内,δ29Si和δ30Si的1σ分别达到0.04‰ (n=8)和0.03‰ (n=6)。当磁场位置过于靠近平台左侧时,测得的δ30Si明显偏低,表明此时可能有少量30Si未完全被法拉第杯接收。而当磁场位置超过29.199时,δ29Si和δ30Si均显著偏负,反映了28Si受到干扰离子的影响。在实际测量中,为了避免仪器质量漂移对同位素比值造成的影响,可将磁场设定在同位素比值稳定的质量范围的中部,即距离信号平台左侧5 milli-amu处。
图3 中分辨条件下平台峰上硅同位素比值稳定范围(29Si和30Si的信号分别放大18.6倍和27倍以方便显示)
Fig.3 Stable mass range of silicon isotope ratio on the plateau peak at medium resolution (the signals of 29Si and 30Si are enlarged by 18.6 and 27 times,
respectively)
3.2.3 数据采集条件 本实验比较了不同积分时间和数据采集次数对29Si/28Si和30Si/28Si内精度的影响。结果显示,采用8 s积分时间时,29Si/28Si和30Si/28Si两组比值的内精度整体上均要优于4 s积分时间(图4a)。并且,在8 s积分时间的条件下,当测试时间达到240 s(即30个数据点)时29Si/28Si和30Si/28Si的内精度(1σ)可达到8 ppm(即8×10
以内。继续增加测试时间,29Si/28Si和30Si/28Si的内精度保持稳定,但30Si/28Si的比值随时间增加逐渐变化(图4b),反映了仪器质量歧视的变化。采用SSB法校正仪器质量歧视,为保证前后两个标准能真实反映样品的质量歧视,需尽快完成整个“标准-样品-标准”测试流程,因而本实验采用8 s积分时间、30次扫描次数的数据采集条件。
3.3 浓度效应
利用SSB测量同位素组成时要求样品与标准溶液中待测元素浓度和基体组成保持一致。为了验证标准和样品之间的浓度不匹配对硅同位素测量的影响,本研究以500 ng/mL NBS28作为标准,分别测量具有不同浓度梯度的GBW04422溶液的同位素组成。结果显示,δ29Si受浓度变化的影响较小,但δ30Si测量值随样品Si浓度增加呈现逐渐上升的趋势(图5)。当样品与标准溶液浓度差异小于20% (0.8≤Csample/Cstandard≤1.2)时,所测δ30Si与标准值的差异不显著;当样品与标准溶液浓度差异超过50%(Csample/Cstandard<0.5或者>1.5)时,δ30Si明显偏离标准值。因此实际分析样品时需确保样品与标准溶液浓度相差不超过20%。
本实验观察到的浓度差异对δ30Si测量影响可能反映了离子流强度变化对空间电荷效应的影响[8]。当硅浓度增加时,等离子体中Si+的电荷密度增大,电荷排斥效应随之增强。因而,高浓度样品中较轻的硅同位素会更偏离离子束的中心,即质量歧视程度增加。然而,也有其它研究发现样品浓度升高会导致所测同位素比值偏低[13~15],或者标准-样品间的浓度不匹配不会造成测量结果产生明显偏差[14]。因而,可以认为浓度效应对同位素测量的影响机理十分复杂,可能与元素性质、测试条件和不同仪器类型等多种因素有关。
3.4 样品酸度和Cl基体
阳离子交换树脂可将金属离子与硅有效分离。实验结果表明,经分离后, 溶液中碱金属Na+和碱土金属Mg2+浓度均低于1 μmol/L。但同时,该步骤会置换树脂上的H+,使其进入流出液中并改变样品酸度。因此,当样品的初始体积改变时,通过离子交换树脂进入到溶液中的H+的量也会改变。Cl
是海水样品中含量最高的阴离子,且实验过程需使用HCl溶解Mg(OH)2沉淀,因此Cl
是分析实际样品时浓度最高的阴离子。为验证溶液酸度和Cl
对硅同位素测量的影响,本研究以Milli-Q水基体中的NBS28溶液为标准,测量HCl浓度为0~1.0 mol/L的GBW04422溶液的硅同位素组成。结果表明, 图6 溶液酸度和Cl基体对硅同位素测试的影响
Fig.6 Impact of acid molarity and matrix of Cl on the determination of silicon isotopes当样品酸度从0增加至1.0 mol/L时,GBW04422标准物质 δ30Si测量值均位于本研究测试的长期稳定性范围内(图6),表明溶液酸度和Cl
基体不会对硅同位素测试造成显著影响。
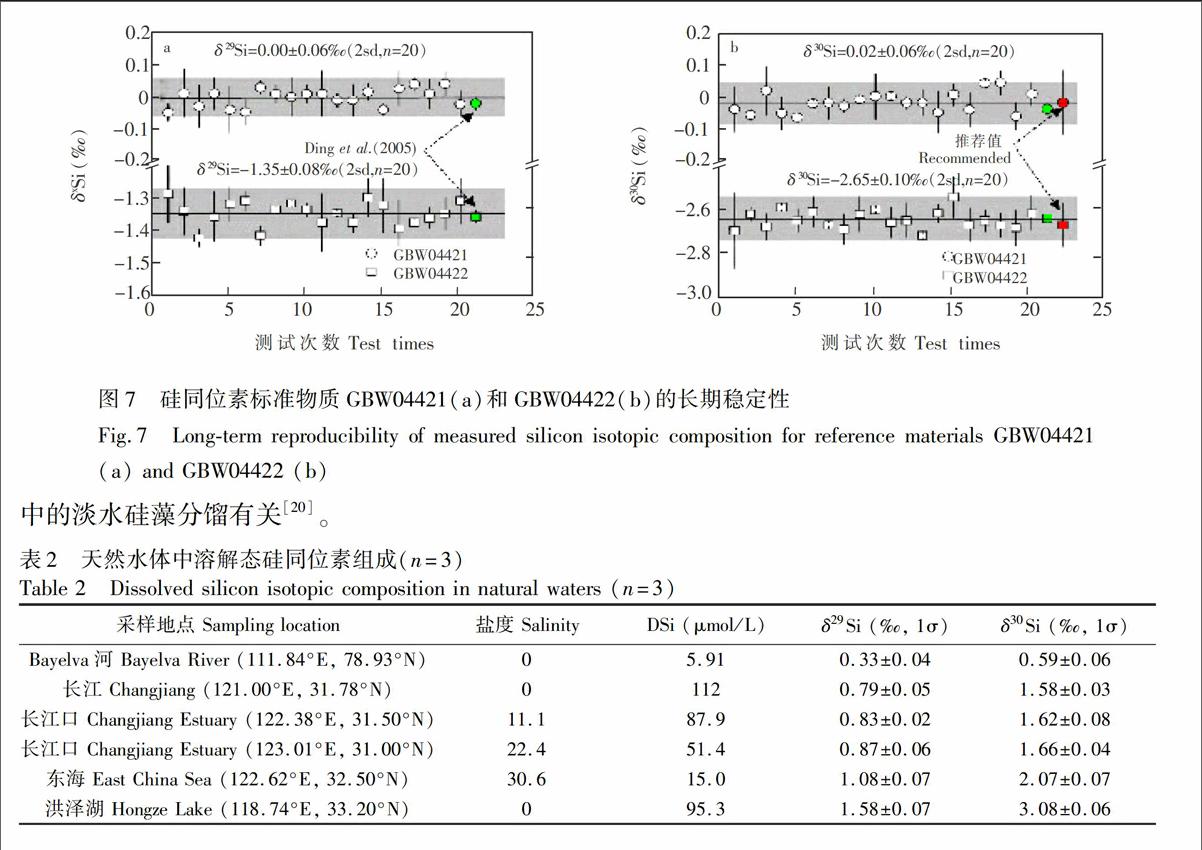
3.5 准确性与长期稳定性
在优化的测试条件下,在 13个月时间内多次测量500 ng/mL GBW04422和GBW04421溶液,并以NBS28作为标准校正仪器质量歧视。GBW04422标准物质δ29Si和δ30Si测定结果分别为
1.35±0.04‰和
2.65±0.05‰(1σ,n=20),GBW04421标准物质δ29Si和δ30Si测定结果分别为0.00±0.03‰和
0.02‰±0.03‰(1σ,n=20)(图7),与推荐值和文献[16]采用气体稳定同位素质谱仪所得结果一致。并且,本研究应用SSB校正方法测试δ29Si和δ30Si所得长期稳定性分别为0.06‰~0.08‰和0.06‰~0.10‰(2σ,n=20),达到文献[9,12,17]报道的应用Mg内标校正相同的精密度(0.08‰~0.14‰)。
3.6 天然水体溶解态硅同位素组成
采用本方法,结合阳离子交换树脂法和单次氢氧化镁共沉淀法,测定了天然水体中溶解态硅同位素组成(表2)。结果表明, 长江水体中DSi浓度和δ29Si、δ30Si均要高于极地地区的Bayelva河,反映了处于温带的长江流域更强的硅酸盐风化和植被的影响[18]。长江口水体中DSi浓度随盐度升高而降低,但硅同位素组成仍接近于长江淡水端元,显示了长江输送的DSi对河口区水体的影响。盐度30以上的东海水体溶解态浓度继续降低,但δ29Si、δ30Si相较于长江口水体出现明显升高,体现了硅藻对DSi的吸收和分馏过程[19]。此外,洪泽湖中δ29Si、δ30Si明显高于长江干流,这可能与水动力条件较弱的湖泊环境中的淡水硅藻分馏有关[20]。
References
1 Berner R A, Lasaga A C, Garrels R M. Am. J. Sci., 1983, 283: 641-683
2 Ragueneau O, Schultes S, Bidle K, Claquin P, Moriceau B. Global Biogeochem. Cycles, 2006, doi: 10.1029/2006GB002688
3 Barnes I, Moore L, Machlan, L, Murphy, T, Shields, W. J. Res. Nat. Bur. Stand. A, 1975, 79: 727-735
4 DING Ti-Ping, WAN De-Fang, LI Jin-Cheng, JIANG Shao-Yong, SONG He-Bin, LI Yan-He, LIU Zhi-Jian. Mineral Deposits, 1988, 7: 90-95
丁悌平, 万德芳, 李金城, 蒋少涌, 宋鹤彬, 李延河, 刘志坚. 矿床地质, 1988, 7: 90-95
5 LI Jin, ZHU Xiang-Kun, TANG Suo-Han. Chinese J. Anal. Chem., 2008, 36(9): 1196-1200
李 津, 朱祥坤, 唐索寒. 分析化学, 2008, 36(9): 1196-1200
6 Engstrm E, Rodushkin I, Baxter D C, hlander B. Anal. Chem., 2006, 78(1): 250-257
7 Cardinal D, Alleman L Y, de Jong J, Ziegler K, André L. J. Anal. At. Spectrom., 2003, 18: 213-218
8 Heumann K G, Gallus S M, Rdlinger G, Vogl J. J. Anal. At. Spectrom., 1998, 13: 1001-1008
9 Georg R B, Reynolds B C, Frank M, Halliday A N. Chem. Geol., 2006, 235: 95-104
10 Zhang A Y, Zhang J, Zhang R F, Xue Y. J. Anal. At. Spectrom., 2014, 12: 2414-2418
11 Grasshoff K, Kremling K. Ehrhardt M.Methods of Seawater Analysis (Third Edition), Weinheim: Wiley: 1999
12 Zambardi T, Poitrasson F. Geostand. Geoanal. Res., 2011, 35, 89-99
13 TANG Ai-Ling, QU Jian-Guo. Chinese J. Anal. Chem., 2013, 41(7):1091-1096
唐爱玲, 瞿建国. 分析化学, 2013, 41(7):1091-1096
14 Teng F Z, Yang W. Rapid Commun. Mass Sp., 2014, 28: 19-24
15 Wang G, Lin Y, Liang X, Liu Y, Xie L, Yang Y, Tu X. J. Anal. At. Spectrom., 2011, 26: 1878-1886
16 Ding T, Wan D, Bai R, Zhang Z, Shen Y, Meng R. Geochim. Cosmochim. Acta, 2005, 69: 5487-5494
17 Abraham K, Opfergelt S, Fripiat F, Cavagna, A J, De Jong J T M, Foley S F, André L, Cardinal D. Geostand. Geoanal. Res., 2008, 32: 193-202
18 Ding T, Wan D, Wang C, Zhang F. Geochim. Cosmochim. Acta, 2004, 68: 205-216
19 de La Rocha C L, Brzezinski M A, DeNiro M J. Geochim. Cosmochim. Acta, 1997, 61: 5051-5056
20 Opfergelt S, Eiriksdottir E S, Burton K W, Einarsson A, Siebert C, Gislason S R, Halliday A N. Earth Planet Sci. Lett., 2011, 305: 73-82
摘 要 建立了应用多接收电感耦合等离子体质谱仪(MC-ICP-MS)测定稳定硅同位素比值的仪器分析方法。在干等离子体、中分辨率条件下,硅同位素高质量端受到C、N、O、H等元素形成的多原子离子的干扰。样品气流量对Si的灵敏度稳定性有重要影响,并且样品气流量增加会导致14N16O的强度增大。中分辨率条件下,低质量数端δ29Si和δ30Si在约9 milli-amu的质量范围内保持稳定,精度优于0.04‰ (1σ)。采用“标准-样品”交叉法正校质量歧视时,为避免浓度效应对硅同位素测试的影响,要求标准和样品之间硅浓度差异低于20%。溶液酸度和Cl基体含量不会对同位素测量造成显著影响。通过优化数据采集参数,29Si/28Si和30Si/28Si的内精度(1σ)可达到8 ppm以内。标准物质长期分析结果显示,δ29Si和δ30Si的长期稳定性可达到0.06‰~0.10‰(2σ,n=20),标准物质GBW04421和GBW04422的测量值与推荐值吻合,表明本方法精确可靠。对淡水(河水、湖水)、半咸水、海水的分析结果表明,应用硅同位素可以示踪天然水体中硅的生物地球化学过程。
关键词 多接收; 硅同位素; 电感耦合等离子体质谱
1 引 言
硅(Si)是地球上丰度仅次于氧的第二大元素,约占地壳总质量的27.6%。自然界中硅循环过程(如硅酸盐风化、硅藻的光合作用)与碳循环紧密相连,在不同时间尺度上影响控制着大气CO2分压和全球气候变化[1,2]。Si具有三个稳定同位素:28Si(92.23%)、29Si(4.67%)、30Si(3.10%)[3]。测定天然水体(如河水、海水、地下水)中溶解态硅同位素组成,可以示踪硅酸盐化学风化、溶解态硅(DSi)生物矿化等硅的生物地球化学循环过程。
最初,硅同位素组成是通过将Si转化为SiF4气体并用稳定同位素质谱仪(IRMS)进行测量[4]。这种方法测量精度高,但氟化过程对实验操作安全要求高,并且所需样品量大,在分析DSi低浓度样品(如表层海水)时受到限制。近年来,多接收电感耦合等离子体质谱法(MC-ICP-MS)的迅速发展使得人们可以利用更少的样品量实现同位素的精确测量。在应用MC-ICP-MS分析硅同位素时,测量结果的精密度和准确度主要受到质谱干扰和仪器质量歧视两个因素的影响[5]。在ICP-MS中,28Si, 29Si和30Si受到来源于溶剂和大气的C、H、O、N几种元素所形成的多原子离子质谱干扰[6],特别是采用湿等离子体进样系统时,高强度的14N16O会导致30Si无法准确测量[7]。ICP-MS的仪器质量歧视主要由空间电荷效应和喷嘴效应导致[8],常用“标准-样品”交叉法(Standard-sample-bracketing,SSB)进行校正。但是,样品基体组成、待测元素浓度变化以及仪器设置上的差异均可引起质量歧视的变化。
本研究系统讨论了应用MC-ICP-MS测定硅同位素比值时多原子离子干扰、仪器参数、浓度效应、样品酸度和基体组成的影响,利用硅同位素标准物质评估了仪器分析精密度和准确度,并测定了典型水体环境中溶解态硅同位素组成。
2 实验部分
2.1 仪器与试剂
NEPTUNE多接收电感耦合等离子体质谱仪(Thermo Fisher Scientific公司);Apex-IR去溶剂装置, 100 μL/min PFA微流同心雾化器(Elemental Scientific, 美国); 硅同位素国际标准物质NBS28; 硅同位素国家标准物质GBW04421和GBW04422(北京地质矿产研究所); 99.99% Na2CO3(国药集团化学试剂有限公司);30%Supra盐酸(Merck, 德国);Milli-Q超纯水(18.2 MΩ·cm,Millipore, 美国);Dowex 50W-X8阳离子交换树脂(Sigma Aldrich公司, 美国)。
硅同位素标准溶液采用碱融法[9]进行制备:准确称取214.8 mg硅同位素标准固体样品和757.1 mg Na2CO3固体,置于铂金坩埚内混匀后950℃熔融1 h;冷却后,加入少量Milli-Q水并稍微加热使其溶解;将溶液转移至容量瓶中,用Milli-Q水润洗坩埚内壁5次,加入10 mL浓HCl,用Milli-Q水定容至100 mL,配制成1000 mg/L标准储备液;储备液转移至LDPE瓶中4℃冷藏保存。硅同位素标准溶液采用Milli-Q水为溶剂逐级稀释至500 ng/mL用于仪器测定。
2.2 样品采集与前处理
天然水体样品(河水、半咸水、海水、湖水)采集后立即用0.4 μm聚碳酸酯膜过滤,滤液置于洁净聚乙烯瓶中,盐酸酸化至pH≈2保存。
DSi>20 μmol/L的淡水样品(湖水、河水)采用阳离子交换树脂法对DSi进行纯化,半咸水、海水以及DSi<20 μmol/L的河水样品采用优化的单次氢氧化镁共沉淀(MAGIC)和阳离子交换树脂法进行富集与纯化,具体实验条件参见文献[10]。DSi浓度采用硅钼蓝比色法[11]测定。
2.3 仪器分析方法
NEPTUNE MC-ICP-MS可在静态模式下同时检测 28Si、29Si和30Si。为降低14N16O的产率,硅同位素采用干等离子体条件进行测试,样品溶液通过100 μL/min PFA雾化器和Apex-IR去溶剂装置(注:未安装膜去溶模块)引入等离子体。每次测试前用含有Si的溶液对进样系统和透镜参数进行优化,以获得最佳精密度和稳定性以及质量分辨率,具体参数设置见表1。
应用MC-ICP-MS测定硅同位素时常采用SSB法和Mg内标法校正仪器的质量歧视。由于Mg和Si的质量歧视程度不完全一致,采用Mg内标法校正质量歧视时通常需要结合SSB法[7,12]。此外,MC-ICP-MS仪器需要通过跳峰才能实现Mg和Si同位素的测定,如此会增加测试时间,减少分析通量。本研究采用SSB法校正仪器质量歧视,硅同位素组成以相对于NBS28的千分差表示,公式如下:
3 结果与讨论
3.1 质谱干扰
干等离子条件下,采用中分辨率扫描Milli-Q水空白溶液,在3个硅同位素高质量数端均存在明显的质谱干扰(图1)。丰度最高的28Si的信号稳定平台宽度约22.6 milli-amu,右侧主要干扰离子是12C16O和14N14N,二者的信号强度均低于20 mV。29Si和30Si的信号稳定平台宽度分别达到33.4和29.2 milli-amu。29Si的主要干扰离子是12C16O1H和14N14N1H,信号强度约1~2 mV,30Si的主要干扰离子为14N16O,信号强度远高于空白溶液中30Si的信号,达到20 mV。14N16O的强度呈现较大波动,可能反映了Apex-IR去除溶剂H2O时的波动。
3.2 仪器参数优化
3.2.1 样品气流量
样品气流量对待测元素灵敏度、信号稳定性和多原子离子产率有重要影响。结果显示,当样品气流量为0.95~0.97 L/min时硅同位素灵敏度和信号稳定性均达到最佳状态(图2a)。过低或者过高的样品气均会导致灵敏度和稳定性有所下降,原因在于样品气过低会造成雾化器反向压力过大,降低样品提升速率和雾化效果的稳定性,而过高的样品气会减小样品在等离子体中的停留时间,影响电离效果。14N16O强度则随着样品气流量增强而持续增加,导致30Si/14N16O的比值不断降低(图2b),这是因为增加样品进样速率时会同时引入更多的溶剂,从而增加氧化物的产率。为确保丰度最低的30Si信号强度达到80 mV左右,同时避免生成过高的14N16O,本研究将样品气流量设在0.96 L/min。
3.2.2 峰平台比值稳定性
图3展示了干等离子条件下扫描含有500 ng/mL Si溶液中硅同位素质谱图。多原子离子干扰对28Si、29Si信号强度影响不大,但14N16O会造成30Si高质量数端信号上升。为准确评估多原子离子贡献对比值的影响以及硅同位素比值稳定的质量范围,以29.192处的比值作为标准,采用SSB法测定峰平台上其它质量数处的同位素组成。结果显示,δ29Si和δ30Si分别在29.188~29.199和29.189~29.198的范围内保持稳定,同位素比值稳定的质量范围达到9 milli-amu(图3)。在比值稳定的质量范围内,δ29Si和δ30Si的1σ分别达到0.04‰ (n=8)和0.03‰ (n=6)。当磁场位置过于靠近平台左侧时,测得的δ30Si明显偏低,表明此时可能有少量30Si未完全被法拉第杯接收。而当磁场位置超过29.199时,δ29Si和δ30Si均显著偏负,反映了28Si受到干扰离子的影响。在实际测量中,为了避免仪器质量漂移对同位素比值造成的影响,可将磁场设定在同位素比值稳定的质量范围的中部,即距离信号平台左侧5 milli-amu处。
图3 中分辨条件下平台峰上硅同位素比值稳定范围(29Si和30Si的信号分别放大18.6倍和27倍以方便显示)
Fig.3 Stable mass range of silicon isotope ratio on the plateau peak at medium resolution (the signals of 29Si and 30Si are enlarged by 18.6 and 27 times,
respectively)
3.2.3 数据采集条件 本实验比较了不同积分时间和数据采集次数对29Si/28Si和30Si/28Si内精度的影响。结果显示,采用8 s积分时间时,29Si/28Si和30Si/28Si两组比值的内精度整体上均要优于4 s积分时间(图4a)。并且,在8 s积分时间的条件下,当测试时间达到240 s(即30个数据点)时29Si/28Si和30Si/28Si的内精度(1σ)可达到8 ppm(即8×10
以内。继续增加测试时间,29Si/28Si和30Si/28Si的内精度保持稳定,但30Si/28Si的比值随时间增加逐渐变化(图4b),反映了仪器质量歧视的变化。采用SSB法校正仪器质量歧视,为保证前后两个标准能真实反映样品的质量歧视,需尽快完成整个“标准-样品-标准”测试流程,因而本实验采用8 s积分时间、30次扫描次数的数据采集条件。
3.3 浓度效应
利用SSB测量同位素组成时要求样品与标准溶液中待测元素浓度和基体组成保持一致。为了验证标准和样品之间的浓度不匹配对硅同位素测量的影响,本研究以500 ng/mL NBS28作为标准,分别测量具有不同浓度梯度的GBW04422溶液的同位素组成。结果显示,δ29Si受浓度变化的影响较小,但δ30Si测量值随样品Si浓度增加呈现逐渐上升的趋势(图5)。当样品与标准溶液浓度差异小于20% (0.8≤Csample/Cstandard≤1.2)时,所测δ30Si与标准值的差异不显著;当样品与标准溶液浓度差异超过50%(Csample/Cstandard<0.5或者>1.5)时,δ30Si明显偏离标准值。因此实际分析样品时需确保样品与标准溶液浓度相差不超过20%。
本实验观察到的浓度差异对δ30Si测量影响可能反映了离子流强度变化对空间电荷效应的影响[8]。当硅浓度增加时,等离子体中Si+的电荷密度增大,电荷排斥效应随之增强。因而,高浓度样品中较轻的硅同位素会更偏离离子束的中心,即质量歧视程度增加。然而,也有其它研究发现样品浓度升高会导致所测同位素比值偏低[13~15],或者标准-样品间的浓度不匹配不会造成测量结果产生明显偏差[14]。因而,可以认为浓度效应对同位素测量的影响机理十分复杂,可能与元素性质、测试条件和不同仪器类型等多种因素有关。
3.4 样品酸度和Cl基体
阳离子交换树脂可将金属离子与硅有效分离。实验结果表明,经分离后, 溶液中碱金属Na+和碱土金属Mg2+浓度均低于1 μmol/L。但同时,该步骤会置换树脂上的H+,使其进入流出液中并改变样品酸度。因此,当样品的初始体积改变时,通过离子交换树脂进入到溶液中的H+的量也会改变。Cl
是海水样品中含量最高的阴离子,且实验过程需使用HCl溶解Mg(OH)2沉淀,因此Cl
是分析实际样品时浓度最高的阴离子。为验证溶液酸度和Cl
对硅同位素测量的影响,本研究以Milli-Q水基体中的NBS28溶液为标准,测量HCl浓度为0~1.0 mol/L的GBW04422溶液的硅同位素组成。结果表明, 图6 溶液酸度和Cl基体对硅同位素测试的影响
Fig.6 Impact of acid molarity and matrix of Cl on the determination of silicon isotopes当样品酸度从0增加至1.0 mol/L时,GBW04422标准物质 δ30Si测量值均位于本研究测试的长期稳定性范围内(图6),表明溶液酸度和Cl
基体不会对硅同位素测试造成显著影响。
3.5 准确性与长期稳定性
在优化的测试条件下,在 13个月时间内多次测量500 ng/mL GBW04422和GBW04421溶液,并以NBS28作为标准校正仪器质量歧视。GBW04422标准物质δ29Si和δ30Si测定结果分别为
1.35±0.04‰和
2.65±0.05‰(1σ,n=20),GBW04421标准物质δ29Si和δ30Si测定结果分别为0.00±0.03‰和
0.02‰±0.03‰(1σ,n=20)(图7),与推荐值和文献[16]采用气体稳定同位素质谱仪所得结果一致。并且,本研究应用SSB校正方法测试δ29Si和δ30Si所得长期稳定性分别为0.06‰~0.08‰和0.06‰~0.10‰(2σ,n=20),达到文献[9,12,17]报道的应用Mg内标校正相同的精密度(0.08‰~0.14‰)。
3.6 天然水体溶解态硅同位素组成
采用本方法,结合阳离子交换树脂法和单次氢氧化镁共沉淀法,测定了天然水体中溶解态硅同位素组成(表2)。结果表明, 长江水体中DSi浓度和δ29Si、δ30Si均要高于极地地区的Bayelva河,反映了处于温带的长江流域更强的硅酸盐风化和植被的影响[18]。长江口水体中DSi浓度随盐度升高而降低,但硅同位素组成仍接近于长江淡水端元,显示了长江输送的DSi对河口区水体的影响。盐度30以上的东海水体溶解态浓度继续降低,但δ29Si、δ30Si相较于长江口水体出现明显升高,体现了硅藻对DSi的吸收和分馏过程[19]。此外,洪泽湖中δ29Si、δ30Si明显高于长江干流,这可能与水动力条件较弱的湖泊环境中的淡水硅藻分馏有关[20]。
References
1 Berner R A, Lasaga A C, Garrels R M. Am. J. Sci., 1983, 283: 641-683
2 Ragueneau O, Schultes S, Bidle K, Claquin P, Moriceau B. Global Biogeochem. Cycles, 2006, doi: 10.1029/2006GB002688
3 Barnes I, Moore L, Machlan, L, Murphy, T, Shields, W. J. Res. Nat. Bur. Stand. A, 1975, 79: 727-735
4 DING Ti-Ping, WAN De-Fang, LI Jin-Cheng, JIANG Shao-Yong, SONG He-Bin, LI Yan-He, LIU Zhi-Jian. Mineral Deposits, 1988, 7: 90-95
丁悌平, 万德芳, 李金城, 蒋少涌, 宋鹤彬, 李延河, 刘志坚. 矿床地质, 1988, 7: 90-95
5 LI Jin, ZHU Xiang-Kun, TANG Suo-Han. Chinese J. Anal. Chem., 2008, 36(9): 1196-1200
李 津, 朱祥坤, 唐索寒. 分析化学, 2008, 36(9): 1196-1200
6 Engstrm E, Rodushkin I, Baxter D C, hlander B. Anal. Chem., 2006, 78(1): 250-257
7 Cardinal D, Alleman L Y, de Jong J, Ziegler K, André L. J. Anal. At. Spectrom., 2003, 18: 213-218
8 Heumann K G, Gallus S M, Rdlinger G, Vogl J. J. Anal. At. Spectrom., 1998, 13: 1001-1008
9 Georg R B, Reynolds B C, Frank M, Halliday A N. Chem. Geol., 2006, 235: 95-104
10 Zhang A Y, Zhang J, Zhang R F, Xue Y. J. Anal. At. Spectrom., 2014, 12: 2414-2418
11 Grasshoff K, Kremling K. Ehrhardt M.Methods of Seawater Analysis (Third Edition), Weinheim: Wiley: 1999
12 Zambardi T, Poitrasson F. Geostand. Geoanal. Res., 2011, 35, 89-99
13 TANG Ai-Ling, QU Jian-Guo. Chinese J. Anal. Chem., 2013, 41(7):1091-1096
唐爱玲, 瞿建国. 分析化学, 2013, 41(7):1091-1096
14 Teng F Z, Yang W. Rapid Commun. Mass Sp., 2014, 28: 19-24
15 Wang G, Lin Y, Liang X, Liu Y, Xie L, Yang Y, Tu X. J. Anal. At. Spectrom., 2011, 26: 1878-1886
16 Ding T, Wan D, Bai R, Zhang Z, Shen Y, Meng R. Geochim. Cosmochim. Acta, 2005, 69: 5487-5494
17 Abraham K, Opfergelt S, Fripiat F, Cavagna, A J, De Jong J T M, Foley S F, André L, Cardinal D. Geostand. Geoanal. Res., 2008, 32: 193-202
18 Ding T, Wan D, Wang C, Zhang F. Geochim. Cosmochim. Acta, 2004, 68: 205-216
19 de La Rocha C L, Brzezinski M A, DeNiro M J. Geochim. Cosmochim. Acta, 1997, 61: 5051-5056
20 Opfergelt S, Eiriksdottir E S, Burton K W, Einarsson A, Siebert C, Gislason S R, Halliday A N. Earth Planet Sci. Lett., 2011, 305: 73-82
