
摘要:目的 建立指紋图谱与一测多评相结合的质量评价模式分析化学转化法富集的甘草药渣总黄酮,为生产中质量控制提供技术支撑。方法 以破壁富集的总黄酮为研究对象建立指纹图谱,以甘草苷为内参物,分别建立异甘草苷、甘草素、异甘草素、甘草酸的相对校正因子,计算其含量。并对计算值与外标法测定值进行比较,以验证一测多评法的实用性与稳定性。结果 建立了甘草药渣总黄酮HPLC指纹图谱,标定了11个共有峰,确定了其中5个共有峰,10批提取物相似度>0.99;一测多评法计算结果与外标法实测值之间相对误差<4%,以多浓度法计算试验所得相对校正因子RSD<2%。结论 本方法准确可靠,专属性强,结果稳定,重复性好,可有效控制甘草药渣提取物的质量。
关键词:指纹图谱;一测多评;甘草药渣提取物;质量评价
DOI:10.3969/j.issn.1005-5304.2018.01.015
中图分类号:R284.1 文献标识码:A 文章编号:1005-5304(2018)01-0069-05
Study on Enrichment of Total Flavonoids from Licorice Residue by Chemical Conversion Method Based on Fingerprint and Quantitative Analysis of Multi-components with
a Single-marker Technique
LIU Xiao-shuan1, XIAO Zheng-guo1, LUO Yan-yan2, LI Xi-xiang1, LI Ji-wen1, BI Ying-yan1, LIU Jun-gang1
1. Pharmacy Department of Gansu Provincial Hospital of Traditional Chinese Medicine, Lanzhou 730050, China;
2. Pharmacy College of Gansu University of Chinese Medicine, Lanzhou 730000, China
Abstract: Objective To establish a combined quality evaluation model of fingerprint and quantitative analysis of multi-components with a single-marker (QAMS) to analyze the total flavonoids from licorice residue by the chemical conversion method; To provide technical support for quality control in production. Methods Total flavonoids of breaking cell wall and enriching were taken as the object of study to establish fingerprint. With liquiritin as internal reference, the relative correction factors of isoliquiritin, glycyrrhizin, isoliquiritigenin and glycyrrhizic acid were established respectively, and the contents were determined. Meanwhile, the calculated values were compared with the measured value by external standard method to verify the practicability and stability of QAMS. Results The HPLC fingerprint of total flavonoids from licorice residue was established. 11 common peaks were identified, and 5 common peaks were identified, and the similarity of the 10 extracts was >0.99; the relative error between the calculated results of QAMS and the measured values of the external standard method was <4%; the RSD of relative correction factor calculated by the multiple concentration method was <2%. Conclusion The method is accurate, reliable, specific, and stable, with good repeatability, which can be used for the quality control of total flavonoids from licorice residue.
Keywords: fingerprint; quantitative analysis of multi-components with a single-marker; licorice residue extract; quality evaluation
中药指纹图谱指中药经适当处理后,采用一定的分析方法得到的能够体现中药整体化学特性的色谱
基金项目:兰州市科技发展计划项目(兰州市科技局2014-2-32)
或光谱图。2015年版《中华人民共和国药典》(一部)新收载特征图谱28个品种,其中提取物3个品种、中药材2个品种、中成药23个品种;新收载指纹图谱9个品种,均为中成药品种[1-3]。目前,指纹图谱已广泛应用到药材的真伪优劣鉴别及新药生产过程的质量控制中[4-6]。对于市场上通过加入单一成分达到现有质量标准的伪药,采用指纹图谱进行多成分的全面控制切实可行。
一测多评方法(QAMS)又称一标多评法,是指在含量测定时,只使用一个对照品,可同时计算出供试品中其他待测成分的含量,使其计算值与实测值符合定量方法学研究的要求。目前,美国药典(USP33)收载的108个植物药标准(13种植物)中36个使用校正因子,欧洲药典(EP7.0)在232个植物药标准中11个使用了一测多评,2015年版《中华人民共和国药典》593个植物药标准(9种植物)通过相对保留时间及对照药材(或对照提取物)的特征图谱确认色谱峰,进而根据相对因子计算其他色谱峰的含量[7],如黄连(小檗碱、表小檗碱、黄连碱、巴马亭含量测定)、丹参、生姜、银杏提取物等。
甘草具有补脾益气、祛痰止咳、缓急止痛、调和诸药之功效。随着对甘草药理作用的进一步认识,全世界对甘草的需求日益增长,甘草资源急剧减少[8]。本课题前期进行了“基于化学转化法破壁富集甘草药渣中总黄酮的开发研究(兰州市科技局人才创新创业项目2014-2-32)”,完成了提取工艺试验及中试研究。本试验以甘草药渣提取物为研究对象,建立指纹图谱,并构建一测多评法测定提取物中5种成分的含量,对甘草药渣提取物进行质量控制。
1 仪器与试药
高效液相色谱仪(包括Waters1525二元泵、Waters717进样器、Waters2487双通道紫外检测器、Breeze工作站);Mettler AE240电子分析天平;SB-3200D型超声波清洗器(宁波新芝生物科技股份有限公司)。
甘草素对照品(批号B-20416)、异甘草素对照品(批号B-21525)、甘草苷对照品(批号B-20414)、异甘草苷对照品(批号B-21524)、甘草酸对照品(B20417),上海源叶生物科技有限公司;甘草饮片(批号150922、150701),甘肃康乐药业有限责任公司,经甘肃省药检院宋平顺主任药师鉴定为Glycyrrhiza uralensis Fisch.的干燥根和根茎;甘草药渣提取物10批(批号分别为20160504、20160505、20160506、20160509、20161102、20161104、20161108、20161107、20161114、20161126),甘肃省中医院制剂室提供,制备过程见文献[9]。无水乙醇(批号20160905)、乙腈(批号20160505)、磷酸(批号20160307)均为色谱纯,天津市大茂化学试剂厂;含量测定用水为娃哈哈纯净水。
2 方法与结果
2.1 定量分析方法验证
2.1.1 色谱条件
色谱柱为岛津Inerisil ODS-3(250 mm×4.6 mm,5 ?m),乙腈为流动相A,0.85%磷酸水溶液为流动相B,梯度洗脱(见表1),柱温25 ℃,检测波长237 nm,流速1 mL/min。
2.1.2 混合对照品的制备
分别精密称取对照品甘草苷5.1 mg、异甘草苷6.6 mg、甘草素5.6 mg、甘草酸5.6 mg、异甘草素5.3 mg,用70%乙醇溶解并定容至25 mL,得到浓度分别为0.204、0.264、0.224、0.224、0.212 mg/mL的单一对照品溶液,分别精密吸取2.0、2.0、0.5、2.0、1.0 mL,置10 mL容量瓶中,用70%乙醇定容,得浓度分别为0.040 8、0.052 8、0.011 2、0.044 8、0.021 2 mg/mL的混合对照品溶液,4 ℃冰箱保存。
2.1.3 供试品溶液的制備
取甘草药渣提取物约100 mg,精密称定,置50 mL锥形瓶中,加70%乙醇至刻度,精密称定,超声处理(180 W,40 kHz)30 min,冷却,称定补足减失的质量,0.45 ?m微孔滤膜过滤,取续滤液,即得。
2.1.4 系统适用性试验
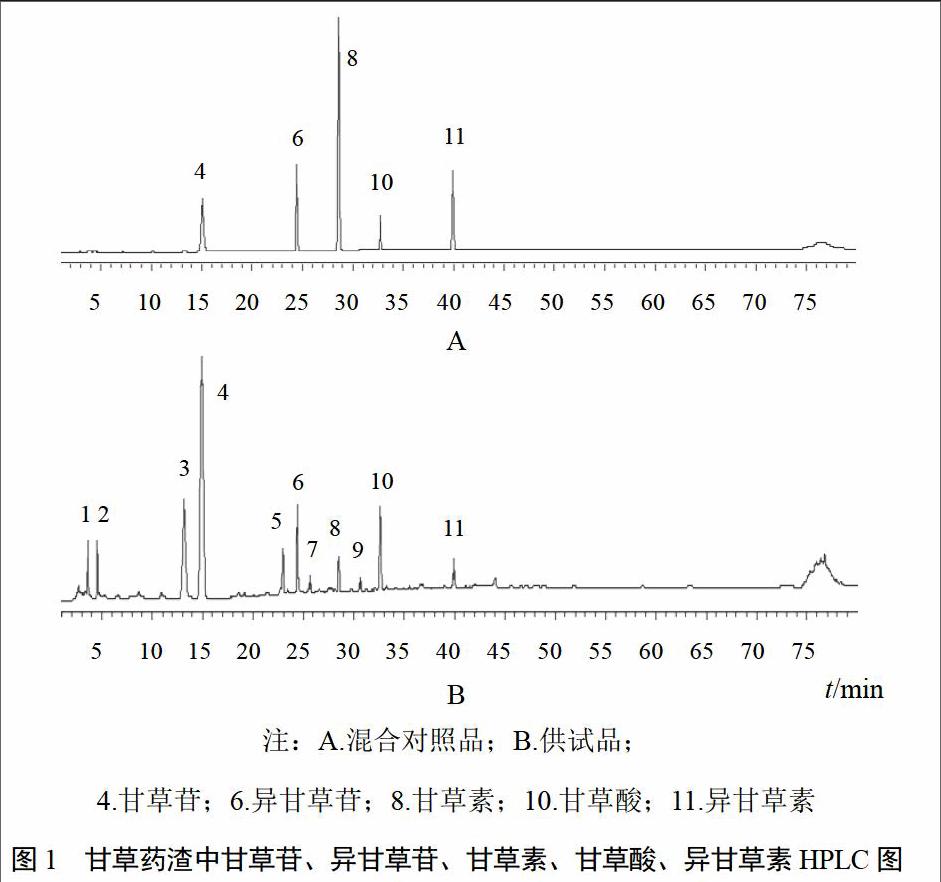
取混合对照品溶液、供试品溶液,按“2.1.1”项色谱条件进样分析,结果各成分色谱峰分离度良好。色谱图见图1。
2.1.5 线性关系考察
分别精密吸取混合对照品溶液0.5、1、5、10、15 ?L进样,在上述色谱条件下记录色谱峰面积,以进样量为横坐标,色谱峰面积为纵坐标,计算回归方程。甘草苷:Y=2 296 046.078X-10 769.500,r=0.999 98,线性范围0.020 4~0.612 0 ?g;异甘草苷:Y=1 361 314.394X-7737.500,r=0.999 91,线性范围0.026 4~0.792 0 ?g;甘草素:Y=5 011 214.286X-15 122.500,r=0.999 83,线性范围0.005 6~0.168 0 ?g;甘草酸:Y=661 750.893X-8479.500,r=0.999 69,线性范围0.022 4~0.672 0 ?g;异甘草素:Y=1 976 024.528X-7840.000,r=0.999 95,线性范围0.010 6~0.318 0 ?g。各成分在线性范围内线性关系良好。
2.1.6 精密度试验
精密吸取同一供试品溶液10 ?L,连续进样6次。结果甘草苷、异甘草苷、甘草素、甘草酸、异甘草素峰面积RSD分别为0.66%、1.52%、0.81%、0.59%、0.71%;以甘草苷(4号峰)为内参物s,其他共有峰相对保留时间为ras=tRa/tRs,RSD分别为0.72%、0.52%、0.40%、0.46%、0.42%,共有峰相似度达1.000。
2.1.7 稳定性试验
取同一批供试品溶液,分别在0、2、4、8、16、24 h进样10 ?L,记录峰面积,RSD分别为0.65%、1.56%、1.85%、0.95%、0.65%,表明供试品溶液在24 h內基本稳定。
2.1.8 重复性试验
取同一批提取物6份,约100 mg,精密称定,按“2.1.3”项下方法制备6份,进样10 ?L,记录峰面积。甘草苷、异甘草苷、甘草素、甘草酸、异甘草素平均含量分别为8.981、2.770、0.401、5.334、0.611 mg/g,峰面积RSD分别为0.68%、1.08%、1.58%、0.90%、1.25%,相似度1.000,符合指纹图谱要求。
2.1.9 加样回收率试验
取已知含量的同一批样品(批号20160506)6份,约60 mg,精密称定,每份加入约1 mL混合对照品溶液,按“2.1.3”项下方法制备供试品溶液,按上述色谱条件进样10 ?L分析。计算甘草苷、异甘草苷、甘草素、甘草酸及异甘草素平均加样回收率分别为100.1%、100.8%、102.2%、103.6、99.2%,RSD分别为1.80%、1.01%、0.85%、1.47%、1.22%。
2.2 指纹图谱分析
2.2.1 指纹图谱的建立
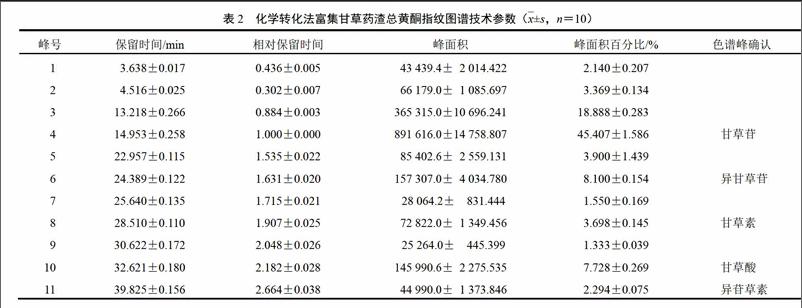
按照中药指纹图谱要求,对10批样品进行检测,其所有成分在80 min内全部出完。采用《中药色谱指纹图谱相似度评价系统》(2012.130723版),将10批样品HPLC图谱数据导入软件,进行相似度分析,生成对照图谱。以甘草苷(4号峰)为参照,11个共有峰的保留时间和峰面积见表2。对照图谱见图2。
2.2.2 相似度评价
以S1样品的指纹图谱作为参照图谱,用平均数法生成10批样品的对照指纹图谱(见图3),10批样品与对照指纹图谱相似度分别为1.000、1.000、1.000、1.000、1.000、0.999、0.998、1.000、0.999、1.000。
2.3 一测多评法分析
2.3.1 5种化合物相对校正因子测定
取“2.1.2”项下混合对照品溶液,分别进样1.0、2.5、5.0、7.5、10.0、15.0 ?L,记录甘草苷、异甘草苷、甘草素、甘草酸、异甘草素的峰面积,以甘草苷为内参物s,采用多浓度计算法,计算相对校正因子。fsi=fs/fi=(As/Cs)/(Ai/Ci)。fs为甘草苷绝对校正因子,fs6、fs8、fs10、fs11分别为异甘草苷、甘草素、甘草酸、异苷草素的相对校正因子。结果见表3。
2.3.2 相对校正因子重现性考察
采用Waters2487考察3种不同型号色谱柱,用岛津Inerisil ODS-3色谱柱考察2种色谱系统对相对校正因子的影响,结果重现性良好,RSD<5%,见表4。
2.3.3 一测多评法与外标法比较
取10批甘草药渣提取物,按“2.1.3”项下方法制备,进样10 ?L测定,分别采用外标法和QAMS法计算5种成分的含量,结果见表5。
3 讨论
甘草有效成分主要为甘草酸和黄酮类化合物。目前对甘草中的甘草酸进行开发、生产的利用率不超过30%,其余则作为废渣丢弃[10-11]。本试验在前期研究基础上建立指纹图谱及一测多评的分析方法,为大规模生产开发甘草药渣提供依据。
试验过程中,选择双通道双波长同时扫描,得到同步双谱图,经比较发现λ237色谱图与λ254色谱图基本接近,但甘草苷在λ237处有最大吸收,保留时间为(14.953±0.258)min。以甘草苷作为内参物,符合易得、稳定、定位准确、重复性好的原则[3],故选择λ237为测定波长。
有文献报道采用变波长法测定[10]。本试验发现变波长测定时背景吸收不易控制、基线不平,且由于特征峰较多,实际操作有困难。本研究最大限度参考药典数据及方法,方便实际操作,耐用性符合要求。
目前已有文献报道从甘草药渣中分离并鉴定了甘草查耳酮等多种成分[12-14],但本试验所用到的样品中尚未发现含有甘草查耳酮B,有待进一步分析考察。
参考文献:
[1] 国家药典委员会.中华人民共和国药典:一部[M].北京:中国医药科技出版社,2015.
[2] 石上梅,于江泳,王旭,等.解读《中国药典》2015年版一部[J].中国药品标准,2015,16(6):403.
[3] 祝明,陈碧莲,石上梅,等.中药指纹图谱技术在中国药典2015年版一部中的应用[J].中国现代应用药学,2016,33(5):661.
[4] LIU B Y, ZHANG C, LV H N, et al. Simultaneous determination of three main analytes of Murraya exotica by HPLC[J]. Journal of Chinese Pharmaceutical Sciences,2015,24(2):88-94.
[5] 宋晓庆,蔡皓,屠鹏飞,等.白芍硫磺熏蒸前后的桂枝汤HPLC特征图谱比较研究[J].中药新药与临床药理,2015,26(1):82-87.
[6] 张倩,霍会霞,顾宇凡,等.沉香药材HPLC-DAD特征图谱研究[J].中国药学杂志,2015,50(3):213-216.
[7] 吴婉莹,果德安.中药整体质量控制标准体系构建的思路与方法[J].中国中药杂志,2014,39(3):351-355.
[8] 王青,苗文娟,向诚,等.乌拉尔甘草化学成分研究[J],中草药,2012, 43(10):1886.
[9] 乔仲和,胡自如.甘草渣中大量黄酮类化合物的分离及甘草的综合开发研究[J].中草药,1997,28(9):522-524.
[10] 刘香南,李明珠,尚晓娜,等.“一测多评”法测定甘草中6种有效成分含量[J].中国实验方剂学杂志,2013,19(24):56-59.
[11] 刘洋,金玉姬,吴湘军,等.甘草黄酮的研究现状及进展[J].吉林医药学院学报,2014,35(2):135-139.
[12] 谢斌,周军辉.甘草渣中甘草总黄酮提取工艺优化研究[J].应用化工, 2016,45(6):1120-1123.
[13] 徐春燕,张娜,韩爱荣,等.甘草渣黄酮回流提取的工艺优化及预处理方法研究[J].食品工业科技,2015,36(2):222-226.
[14] 廉宜君,刘红,马彦梅,等.甘草渣多糖的大孔树脂分离纯化及抗氧化活性的研究[J].石河子大学学报:自然科学版,2015,33(3):351-356.
(收稿日期:2017-03-23)
(修回日期:2017-04-11;编辑:陈静)
- 地铁信号系统的仿真研究
- 微探架空输电线路覆冰在线监测装置及防护措施
- 基于iOS的无人机上位机软件设计
- 浅谈公路桥梁隧道工程施工防水设施应用
- 高压电气设备在线检测技术的探讨
- 大数据技术在企业业财融合中的应用探索
- 超双疏钻井液在大港油田六间房区块房29-2-1L井应用
- 浅谈神朔铁路交流机车司机室冬季防寒的措施
- 汽车零部件的需求预测与库存策略应用研究
- 浅谈微喷灌溉技术在蔬菜种植上的应用
- 试论轧钢技术的分类及在生产中的应用
- 超声波测量钻井卡点的方法研究
- 轧钢加热炉在生产中的温度控制研究
- 浅析聚乙烯装置后部造粒对粒料颜色的影响
- 互联网技术与会计专业课程体系建设深度融合的路径研究
- 浅谈人才战略新思维
- 基于碳优化的农产品冷链物流体系研究
- 企业财务报表分析中常见问题及方案初探
- 试论管理会计与财务会计在企业财务管理中的结合
- 浅谈会计信息化对企业财务管理的影响及对策
- 负债融资对我国生物医药上市公司研发投入的影响研究
- PPP项目财务管理与会计核算分析
- 关于房地产项目公司控制权转移的财务思考
- 做好医院内部审计工作的几点思考
- 农村金融服务体系的问题及完善探究
- subarticulations
- subarticulative
- sub-assembly
- subassociation
- subassociational
- subassociations
- subattorney
- subattorneys
- subattorneyship
- subattorneyships
- subaudibilities
- subaudibility
- subaudibleness
- subaudiblenesses
- subaudibly
- subauditor
- subauditors
- subautomatic
- subautomatically
- subaveragely
- subbailiff
- subbailiffs
- subballast
- subballasts
- sub-basal
- 婆媳两个生孩子——老少都喜欢
- 婆媳争吵
- 婆媳二人守寡——没公夫
- 婆媳二人守寡——没公(功)夫
- 婆媳亲,全家和
- 婆媳吵架——各有各的理
- 婆媳吵架儿子劝
- 婆媳吵架儿子劝——左右为难
- 婆媳和睦,全家幸福;婆媳斗,全家愁
- 婆嫂船
- 婆子
- 婆子烧庵
- 婆家
- 婆心
- 婆心苦口
- 婆欢喜
- 婆母
- 婆然
- 婆猴伎
- 婆猴技
- 婆珊婆演底
- 婆留
- 婆罗州
- 婆罗洲
- 婆罗门