郭丽蓉 徐松 吴银双 李娜
[摘要]目的 建立测定酚氨咖敏颗粒中四种有效成分溶出的方法。方法 溶出试验采用桨法,转速为50 r/min,溶出介质体积为900 ml,选取0.1N盐酸、pH6.8磷酸盐缓冲液、水及pH4.0醋酸盐缓冲液为溶出介质。结果 咖啡因、对乙酰氨基酚、马来酸氯苯那敏及氨基比林等四种有效成分在1 h内均能溶出标示量的85%以上。结论 本研究为酚氨咖敏颗粒的体外溶出曲线一致性评价提供了试验依据。
[关键词]对乙酰氨基酚;氨基比林;咖啡因;马来酸氯苯那敏;溶出曲线
[中图分类号] R973.2? ? ? ? ? [文献标识码] A? ? ? ? ? [文章编号] 1674-4721(2020)5(b)-0015-06
Study on the dissolution of four effective components in Paracetamol, Aminophenazone Phenacetin, Caffeine and Chlorphenamine Maleate Granules
GUO Li-rong? ?XU Song? ?WU Yin-shuang? ?LI Na
Yunnan Institute of Materia Medica, Yunnan Baiyao Group Innovation and R & D Center, Yunnan Province Company Key Laboratory for Traditional Chinese Medicine and Ethnic Drug of New Drug Creation, Kunming? ?650111, China
[Abstract] Objective To establish a method for the dissolution of four effective components in Paracetamol, Aminophenazone Phenacetin, Caffeine and Chlorphenamine Maleate Granules. Methods The paddle method was adopted, the rotation rate was 50 r/min, and the volume of dissolution mediums was 900 ml. Hydrochloric acid solution (0.1N), phosphate buffer (pH=6.8), water and acetic acid-sodium acetate buffer solution (pH=4.0) were adopted as dissolution mediums. Results The dissolution of Caffeine, Paracetamol, Chlorpheniramine Maleate and Aminopyrine were all above 85% of the labelled quantity in 1 h. Conclusion The study provides the experimental basis for the consistency evaluation of the dissolution curves of Paracetamol, Aminophenazone Phenacetin, Caffeine and Chlorphenamine Maleate Granules in vitro.
[Key words] Paracetamol; Aminopyrine; Caffeine; Chlorphenamine Maleate; Dissolution curve
目前酚氨咖敏顆粒的国家药品标准[1]采用高效液相色谱法,用2种色谱条件对氨基比林、咖啡因及马来酸氯苯那敏等三个组分的含量进行测定,未收载对乙酰氨基酚的含量控制方法。且该品种为口服固体制剂,溶出度是制剂评价的重点项目,体外溶出度试验是评价口服固体制剂内在质量的一种重要方法[2],尤其是多种pH溶出介质中的溶出行为,不仅可反映仿制制剂内在品质是否一致,还可提高药物生物等效性试验的成功率[3-7]。建立有区分力的溶出曲线可以在一定程度上比较不同制剂的差别,评价药品的质量[8-9]。故本研究根据国家颁布的相关指导原则规定,建立四组分的含量测定方法及溶出曲线测定方法,探索酚氨咖敏颗粒中四种有效成分的溶出情况,为仿制制剂的生产工艺和内在质量提升研究提供参考及思路[10]。
1仪器与试药
1.1仪器
Waters 2695高效液相色谱仪(美国 Waters);Waters 2998 PDA二极管阵列检测器(美国 Waters);Waters XTERRA MS C18(4.6×250 mm,5 μm);Agilent 1260高效液相色谱仪(美国安捷伦);Agilent DAD二极管阵列检测器(美国安捷伦);Agilet ZORBAX XDB C18(4.6×250 mm,5 μm)。
1.2试药
酚氨咖敏颗粒(H5321827,批号190701、190702、190703);对乙酰氨基酚对照品(100018-20161,中国食品药品检定研究院);氨基比林对照品(10053-201302,中国食品药品检定研究院);咖啡因对照品(171215-201512,中国食品药品检定研究院);马来酸氯苯那敏对照品(100047-201507,中国食品药品检定研究院)。
2方法与结果
2.1色谱条件
2.1.1氨基比林、对乙酰氨基酚及咖啡因检测的色谱条件
色谱柱为Waters XTERRA MS C18(4.6×250 mm,5 μm);流动相为甲醇-乙腈-水(35∶5∶60);流速为1.0 ml/min;检测波长为273 nm。
2.1.2马来酸氯苯那敏检测的色谱条件
色谱柱为Agilet ZORBAX XDB C18(4.6×250 mm, 5 μm);流动相为乙腈-0.3%十二烷基磺酸钠溶液-磷酸=60∶40∶0.02(用三乙胺调pH到3.3);流速为1.0 ml/min;检测波长为215 nm。
2.2方法学验证
2.2.1氨基比林、对乙酰氨基酚及咖啡因含量方法学验证
2.2.1.1专属性? 取流动相作为空白对照溶液;按处方比例称取各辅料,加流动相溶解,制成空白辅料溶液;酚氨咖敏颗粒研细,取细粉300.17 mg(约相当于氨基比林10 mg),加入流动相25 ml,称定重量,超声处理30 min,放置至室温,用流动相补足重量,滤过,精密量取续滤液5 ml,置50 ml量瓶中,用流动相定容至刻度,摇匀,过0.45 μm滤膜,作为供试品溶液。
取上述各溶液10 μl,采用高效液相色谱法检测,结果见图1~3,提示该方法专属性良好。
2.2.1.2精密度? 精密移取三种对照品溶液各5 ml,置于50 ml量瓶中,加流动相定容至刻度,得混标溶液。取混标溶液10 μl,重复进样6次,计算峰面积的RSD值。结果提示,对乙酰氨基酚峰面积的RSD为0.31%,咖啡因峰面积的RSD为0.48%,氨基比林峰面积的RSD为0.49%,符合验证要求(RSD≤1.0%),提示该方法精密度良好。
2.2.1.3重复性? 平行制备6份供试品溶液,各进样10 μl,计算各成份峰面积RSD值。结果提示,对乙酰氨基酚峰面积的RSD为1.51%,咖啡因峰面积RSD为1.51%,氨基比林峰面积的RSD为1.51%,重复性符合验证要求(RSD≤2.0%)。
2.2.1.4线性? 精密称取对乙酰氨基酚对照品60.45 mg、咖啡因对照品12.51 mg和氨基比林对照品40.28 mg,置于100 ml量瓶中,加流动相溶解定容至刻度,作为混标储备液。分别精密量取混标儲备液0.5、1、2、5、6、8 ml于50 ml量瓶中,加流动相定容至刻度,各进样10 μl,记录色谱图。以浓度为横坐标,峰面积为纵坐标绘制线性关系图,结果见图4~6。结果提示,对乙酰氨基酚浓度在6.04~96.72 μg/ml内,咖啡因浓度在1.25~20.02 μg/ml内,氨基比林浓度在4.03~64.45 μg/ml内,线性符合验证要求(r≥0.998)。
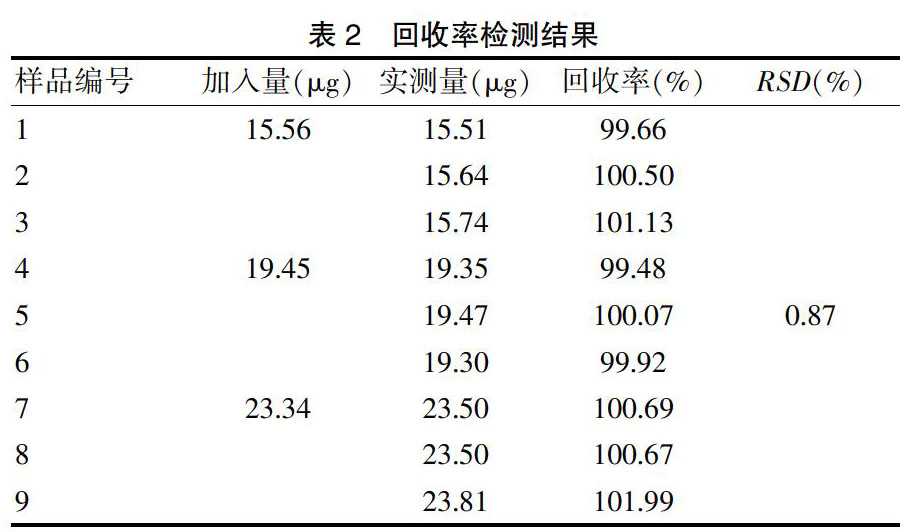
2.2.1.5回收率? 分别精密量取混标储备液4、5、6 ml于50 ml容量瓶中,各加入空白辅料溶液5 ml,最后加流动相定容至刻度。平行制备3份,各进样10 μl,计算9份样品回收率的RSD,结果见表1,结果提示,该方法回收率良好。
2.2马来酸氯苯那敏含量测定方法学验证
2.2.1专属性
取酚氨咖敏颗粒约1 g于三角瓶中,加50 ml流动相后称重,超声处理10 min,放置至室温,称重,用流动相补足损失,过0.22 μm滤膜,取续滤液,进样20 μl,记录色谱图(图7),结果提示,方法专属性良好。
2.2.2精密度
取同一份供试品溶液,连续进样6次,记录峰面积。结果显示,峰面积RSD为0.32%,提示该方法精密度良好。
2.2.3重复性
平行制备6份供试品溶液,各进样10 μl,记录峰面积。结果显示,峰面积的RSD为0.75%,提示该方法的重复性良好。
2.2.4线性
精密称取马来酸氯苯那敏对照品适量,加流动相制成浓度约为0.21 mg/ml的线性储备溶液。取线性储备液,逐级稀释制成浓度为21.24、8.50、4.25、2.12和0.85 μg/ml的线性溶液。
取上述线性储备液和线性溶液,各进样10 μl,记录峰面积。以浓度为横坐标,峰面积为纵坐标绘制线性关系图,结果见图8,提示马来酸氯苯那敏在浓度0.85~21.24 μg/ml内,线性关系良好。
2.2.5回收率
按处方比例加入各辅料,配制成空白辅料储备液。精密称取马来酸氯苯那敏对照品适量,加流动相制成浓度为0.1951 mg/ml的马来酸氯苯那敏对照品溶液;分别取马来酸氯苯那敏对照品溶液0.8、1.0及1.2 ml各3份于10 ml量瓶中,然后分别加入空白辅料储备液各5 ml,用流动相定容至刻度,摇匀,微孔滤膜过滤,取续滤液进样检测,记录峰面积,计算回收率,结果见表2,提示该方法回收率良好。
2.3四种主成分溶出研究
2.3.1试验条件
根据2015年版《中国药典》[11]及《普通口服固体制剂溶出度试验技术指导原则》分别配制溶出介质。介质体积为900 ml,37℃采用桨法,转速50 r/min,每次取样10 ml后补充相应的介质10 ml,取样时间为5、10、15、20、30、45及60 min。样品经0.45 μm滤膜过滤后,采用高效液相色谱法检测,按照公式计算累积溶出度。
2.3.2溶出结果
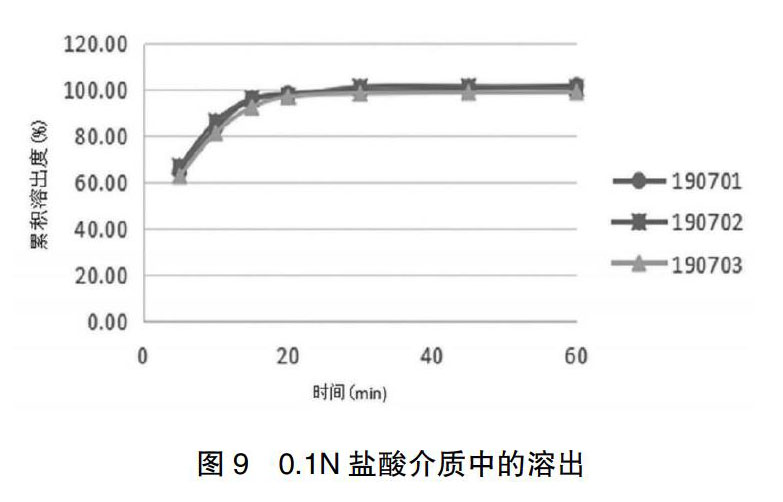
2.3.2.1酚氨咖敏颗粒中对乙酰氨基酚在四种介质中的溶出情况? 酚氨咖敏颗粒中对乙酰氨基酚在四种介质中的溶出曲线见图9~12。
2.3.2.2酚氨咖敏颗粒中氨基比林在四种介质中的溶出情况? 酚氨咖敏颗粒中氨基比林在四种介质中的溶出曲线见图13~16。
- 知行合一:让学生在道德与法治课程中真正成长
- 班级管理:做学生的导航员
- 核心素养下构建高中物理高效课堂的研究
- 中小学情思音乐教学初探
- 引导深度学习提升初中数学素养
- 试论初中体育教学中的快乐田径课堂构建
- 浅析初中数学课堂教学中学生自主学习能力的培养
- 深度学习背景下初中化学高效课堂的构建
- 培养高中学生历史学科核心素养的有效途径和方法
- 高中语文教学应注重生涯规划教育的渗透
- 浅谈如何在体育课中构建师生亲情关系
- 中学教学发展性评价实施策略
- 浅谈高中生物教学
- 疫情防控期间在线语文课堂教学的细节处理
- 高中生物实验教学中学生能力的培养
- 初中语文教学中进行生命教育的实践与思考
- 情景游戏在幼儿教育中的应用探究
- 基于英语学习活动观的初中英语过程性写作教学策略探究
- 积极德育视野下高中生精神成长实践研究
- 在初中历史教学中培养学生的爱国主义
- 言语秒天下,情感抒自然
- 在语文课堂中渗透中国传统“孝”文化
- 基于核心素养的中学生情志生命教育措施的行动研究
- 优化高中政治教学的有效途径
- 初中地理情境教学策略探究
- proacceptance
- proacquisition
- proacquittal
- proacting
- proaction
- proactivator
- proactive
- proactively
- proactive marketing
- proactivemarketing
- proactivenesses
- proactiveness, proactivity
- proactives
- proactivities
- proadministration
- proadmission
- proadoption
- proadvertising
- proadvertizing
- proagitation
- proagrarian
- proagreement
- proairplane
- proalien
- proalliance
- 余儿
- 余光
- 余光分人
- 余党
- 余兴
- 余兴未消
- 余兵
- 余再续申
- 余冠英
- 余冻
- 余凛
- 余刃
- 余刃恢恢
- 余分
- 余切
- 余利
- 余制
- 余剩
- 余剩未尽的威力
- 余割
- 余力
- 余务
- 余势
- 余勇
- 余勇可贾