邹婷婷 杨金易 徐振林 王弘 孙远明 雷红涛 谭学才 沈玉栋
摘 要:采用共有骨架结构结合不饱和线性手臂半抗原策略,通过免疫新西兰大白兔获得了可同时识别他达拉非、氨基他达拉非和去甲基他达拉非的特异性多克隆抗体,通过优化反应条件,建立了微孔侧流免疫层析方法,用于保健酒及口服液样品中非法添加的他达拉非类药物残留的快速检测。本方法定量检出限为0.05~0.30 ng/mL, 裸眼消线值在50~100 ng/mL之间,检测时间小于10 min;与其它结构功能类似物无明显交叉反应,特异性良好。对保健酒及口服液样品的添加回收率在78.7%~117.8%之间,相对标准偏差小于15%。本方法检测结果与HPLCMS/MS法检测结果一致,适用于保健酒及口服液中他达拉非类药物的快速筛查。
关键词:他达拉非; 微孔侧流免疫层析; 保健酒及口服液; 半抗原; 多克隆抗体1 引 言
他达拉非(Tadalafil)是一种5型磷酸二酯酶(Phosphodiesterase 5, PDE5)抑制剂,具有治疗男性阴茎勃起功能障碍(Erectile dysfunction, ED)的作用[1],作为处方药已在多个国家上市。但他达拉非类药物对人体的心血管、消化和神经系统存在一定副作用,特别在不知情的情况下,与硝酸酯类药物同时食用,会对食用者健康和生命造成严重威胁[2]。因此,2012年国家食品药品监督管理总局发布了“食药监办保化\[2012\]33号”文件,明确禁止在保健品中添加他达拉非、氨基他达拉非PDE5抑制剂。然而,利益的驱动导致壮阳抗疲劳类保健品中非法添加他达拉非类药物现象仍屡有发生[3,4],且为了规避执法检查,还推出了他达拉非结构类似物—去甲基他达拉非[5]。因此,建立他达拉非类药物多残留检测方法具有重要的实际意义。
目前,他达拉非类药物非法添加的检测方法主要为实验室大型仪器确证技术,如高效液相色谱法[6,7]、液相色谱质谱联用法[8]、气相色谱质谱联用[9]等。 但单一大型仪器确证技术难以对食品进行高效的监测, 而基于抗原与抗体免疫识别的快速筛查方法作为与之互补的检测技术,已成为食品安全高效检测的不可或缺主流快速检测手段。微孔侧流免疫层析技术(Microwell lateral flow immunochromatography, mwLFIA)作为一种新型免疫层析技术,具有灵敏度高、操作简便、耗时短的优点。该方法是对传统免疫层析技术的改进,其原理是:先使游离的待测物和标记抗体在微孔中均相溶解后充分反应,然后再通过层析至固相载体上的包被抗原进行快速捕获金标抗体而聚集显色,微孔中游离待测物和金标抗体反应越充分,游离的金标抗体越少,固相载体上包被原捕获的金标复合物越少,显色就越浅,检测灵敏度越高。该技术作为仪器确证法的快速筛查互补方法,引起研究者的广泛关注。基于该原理,研究者已经建立了牛奶基质中喹诺酮类药物和苯并咪唑类化合物的多残留微孔侧流免疫层析检测技术[10,11],检出限分别为0.1 和0.77 ng/mL,灵敏度高,适用于实际样品的检测。然而,目前同时检测他达拉非类药物的mwLFIA方法还未见报道。
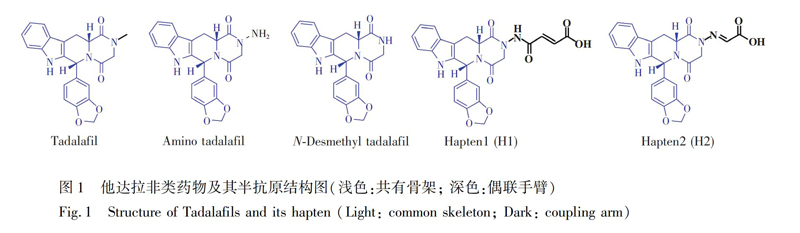
本研究采用共有骨架结构结合不饱和线性手臂半抗原策略[12](图1),最大限度保留他达拉非骨架结构,通过免疫新西兰大白兔,制备可同时识别他达拉非、氨基他达拉非和去甲基他达拉的非特异性多克隆抗体,并基于微孔侧流免疫层析技术,建立了可检测他达拉非类药物的侧流免疫分析方法,为食品安全监测提供了技术支持。
2.1 仪器与试剂
U3010紫外可见光谱仪(日本 Hitachi 公司); HM3030 XYZ三维划膜喷金仪和CTD300数控裁条机(上海金标生物科技有限公司); MD100金标荧光二合一免疫检测仪(南京微测生物科技有限公司); AB SCIEX 5500三重四极杆质谱仪(美国AB SCIEX 公司); 5417R高速离心机(德国 Eppendorf 公司); NEVAP氮吹仪(德祥科技有限公司)。
半抗原H1、H2、20 nm金纳米粒子(Au nanoparticles, AuNPs)溶液(本实验室自制); 马来酸酐、他达拉非、氨基他达拉非、去甲基他达拉非及西地那非类药物(上海阿拉丁试剂有限公司); 羊抗兔IgG抗体、牛血清蛋白(BSA)、卵清蛋白(OVA)、钥孔血蓝蛋白(KLH)(美國Sigma公司); 硝酸纤维素膜、DB6吸水纸(上海杰一生物科技有限公司); H8底板(基因有限公司); 玻璃纤维膜(美国Ahlstrom公司); 样品垫处理液和金标复溶液(广州万联生物科技有限公司); 其它试剂购自广州化学试剂公司。保健酒和口服液购自广州市某药店。

实验中所用的溶液:0.1 mol/L K2CO3溶液(13.8 g/L),10% BSA(10 g/L BSA),包被液(pH 9.6,0.1 mol/L 碳酸盐缓冲液),0.1 mol/L 磷酸盐缓冲溶液(PB,3.117 g/L KH2PO4, 20.742 g/L Na2HPO4·12H2O), 0.1 mol/L KOH溶液(5.6 g/L)。
2.2 实验方法
2.2.1 半抗原的合成和鉴定 按照文献\[13\]方法制备半抗原H1。半抗原H2的制备方法如下: 在25 mL两口圆底烧瓶中,将117.12 mg氨基他达拉非溶于5 mL甲醇,逐滴加入溶有37.02 mg乙醛酸的甲醇(3 mL),60℃回流反应3 h后停止加热,常温下继续搅拌至白色固体析出,真空抽滤,用2 mL甲醇洗涤滤饼2次,滤渣烘干,即得白色固体状半抗原H2,经ESIMS质谱鉴定合成成功(ESIMS:m/z 445.63 \[M\])。
2.2.2 人工抗原的合成与鉴定 采用活泼酯法[14,15]将半抗原H1与BSA偶联作为免疫原(H1BSA),将H1和H2分别与载体蛋白OVA、KLH偶联得到包被原(H1OVA、H1KLH、H2OVA、H2KLH),采用紫外扫描法并结合免疫血清的效价与抑制实验进行鉴定[14,15],分装于离心管中,20℃冻存。
2.2.3 抗体的制备与纯化 参照文献\[16\],采用免疫原H1BSA免疫3只2.0~2.5 kg雌性新西兰大白兔,制备多克隆抗体。通过酶联免疫吸附分析法(Enzymelinked immunosorbent assay, ELISA)测定抗血清效价与抑制率,进行抗体性能鉴定。采用辛酸硫酸铵沉淀法[17]纯化抗体,于20℃冻存。
2.2.4 包被原/抗体组合的筛选
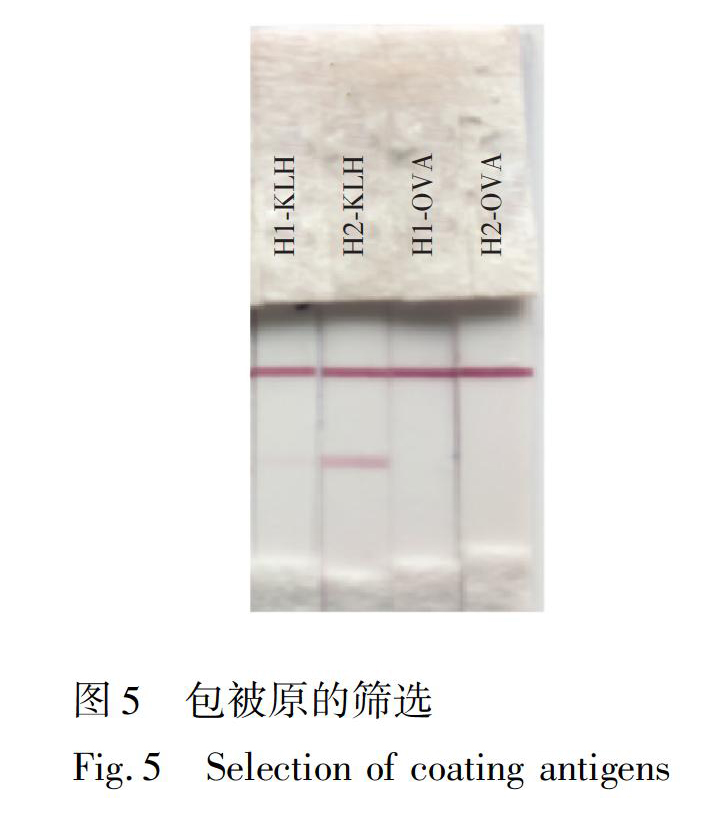
分别以H1OVA、H1KLH、H2OVA、H2KLH为包被原,采用间接竞争ELISA方法,考察抗体/包被原组合效价和抑制,并结合试纸条质控线(Control line, C line)与检测线(Test line, T line)显色情况确定最优抗体/包被原组合,进行mwLFIA方法优化。
2.2.5 AuNPs溶液pH值优化 采用文献\[18\]的方法制备和鉴定AuNPs溶液。在4个装有1 mL AuNPs溶液的离心管中,用0.1 mol/L K2CO3溶液调节pH值分别为8.0、8.5、9.0和9.5,各加入适量抗体进行标记,振荡10 min,再加入20 μL 10% BSA,振荡20 min,4℃ 12000 r/min离心15 min,弃上清液,沉淀加入等体积溶液振荡复溶,裸眼观察复溶情况,测定各复溶液在525 nm处的吸光值,将易复溶、吸光值最高的复溶液的pH值作为最佳标记pH值。
2.2.6 标记抗体浓度优化 在已确定pH值的AuNPs溶液中加入适量抗体(8.0 mg/mL),终浓度分别为4.0、8.0 、12.0、16.0、20.0和24.0 μg/mL,振荡孵育10 min后,加入20 μL 10% BSA溶液,继续振荡20 min,离心后弃上清液,等体积振荡复溶, 测定525 nm吸光值,选择维持AuNPs溶液为红色的最低抗体浓度为最佳抗体标记浓度[19]。
2.2.7 金标抗体用量和包被原浓度的选择
分别吸取2.2.6节制备的金标抗体溶液3、6和9 μL于微孔板中,37℃烘干; 将包被原分别以2.0和4.0 mg/mL划膜于试纸条,烘干后,通过棋盘法检测阴性缓冲液(0.1 mol/L PB溶液)和50 ng/mL他达拉非阳性标准液,用免疫层析读数仪测定,选择阴性孔试纸条T/C≈1.0,T、C线显色明显,且阳性抑制率高的作为最佳金标抗体量/包被原浓度组合。
2.2.8 试纸条的组装和金标微孔的制备
试纸条由样品垫、硝酸纤维素膜(NC膜)、吸水纸和PVC底板四部分组成,NC膜上T线为包被原,C线为羊抗兔IgG抗体,将样品垫、NC膜和吸水纸依次衔接粘贴在PVC底板上,切成3.05 mm宽试纸条(图2),密封干燥保存。吸取按2.2.7节确定的金标抗体量铺于96孔板的微孔,振荡均匀,37℃烘干过夜,密封干燥保存。
2.2.9 测定步骤及結果判定 吸取80 μL缓冲液于金标微孔内,室温孵育5 min后,将试纸条垂直插入微孔,反应6 min。裸眼观察判断: T线和C线显色相近时,结果判定为阴性; T线颜色浅于C线时,结果判定为弱阳性; T线无色结果判定为强阳性; C线无色,试纸条判定为失效(图2)。同时,采用免疫层析读数仪读数, 建立标准曲线, 并确定样本中他达拉非及其类似物含量。
2.2.10 样品前处理 准确量取0.5 mL保健酒或口服液样品于10 mL离心管,加入2 mL KOH溶液(0.1 mol/L)后, 用2 mL CHCl3振荡萃取40 s,静置分层后, 取下清液于10 mL离心管,重复萃取2次,合并下清液,50℃氮吹至干,用50 μL 0.1%乙酸甲醇溶液复溶后,加0.1 mol/L PB稀释至0.5 mL。3 结果与讨论
3.1 AuNPs制备与鉴定
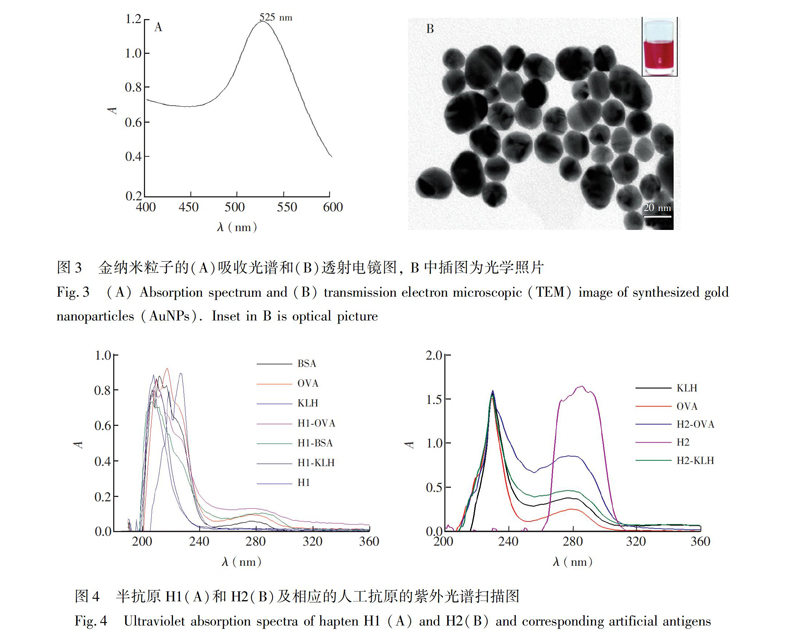
通过紫外扫描(图3A)可知,该AuNPs溶液的最大吸收波长为525 nm, 最大吸收峰的峰宽较窄,说明制备的AuNPs颗粒均匀,通过透射电镜(图3B)对其进行扫描,计算100个粒子, 得AuNPs的平均粒径为20 nm。通过裸眼观察(图3B插图), 制备的AuNPs色泽鲜艳,澄清透明。
3.2 人工抗原的合成与鉴定
以半抗原及载体蛋白为参照对人工抗原进行紫外光谱扫描(图4),发现人工抗原的吸收光谱与半抗原和载体蛋白均有所不同,特征吸收峰位移或峰型发生一定变化,说明半抗原共价偶联至载体蛋白表面。
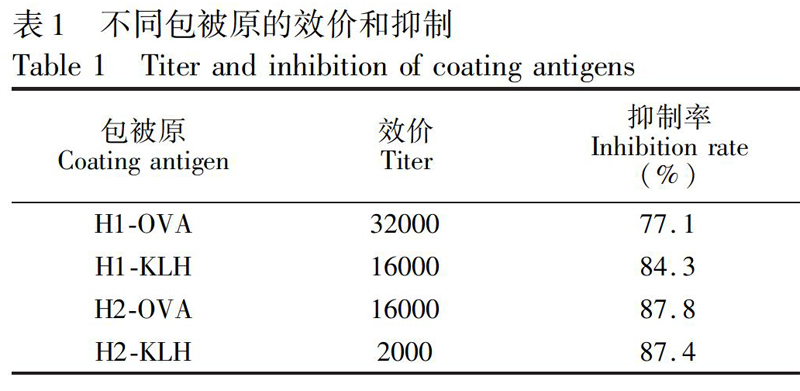
后续抗血清竞争抑制实验显示针对药物和半抗原显现出特异性识别反应(表1),说明产生了特异性抗体,从而证明人工抗原合成成功。
3.3 包被原的筛选
采用间接竞争ELISA方法,以他达拉非(1 μg/mL)为竞争物,以同源和异源包被的方式测定纯化后抗体的效价和抑制率,结果见表1,对于所有包被原,抗体H1BSA均显示出较好的亲和力和识别效果。进一步将包被原以1 mg/mL的浓度通过划膜包被于NC膜的检测线上。
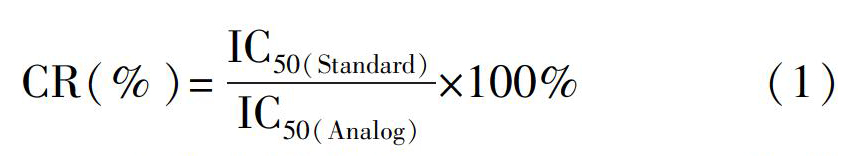
3.4 金标抗体pH值与抗体浓度的确定
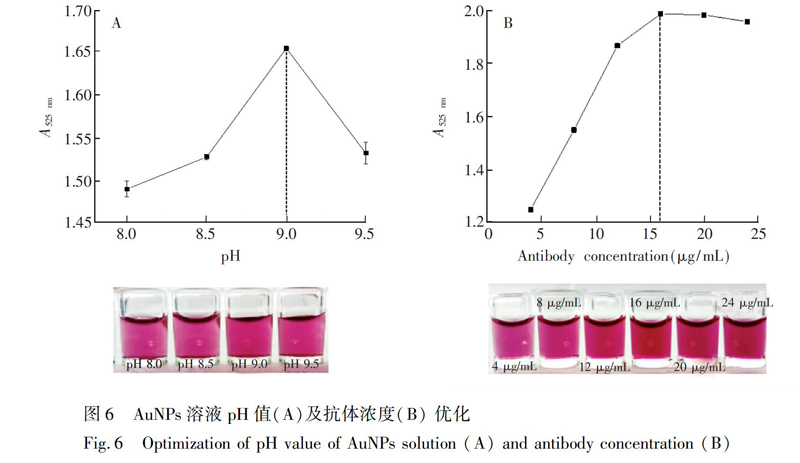
在不同pH值的标记条件下制备金标抗体(图6A),发现随着AuNPs溶液pH值增大,其吸光值先变大后变小,当pH=9.0时,吸光值最大,此时AuNPs溶液处于稳定状态。因此,选择制备金标抗体的最佳pH=9.0。同时,在不同抗体浓度下制备金标抗体(图6B),发现当抗体浓度为16.0 μg/mL时,AuNPs溶液颜色和525 nm处的吸光值开始趋向平缓,因此制备金标抗体时选择抗体浓度为16.0 μg/mL。
3.5 金标抗体用量和包被原浓度的确定
采用棋盘法确定最佳抗体和包被原浓度,结果表明,金标抗体(16.0 μg/mL)用量为9 μL、包被原浓度为4 mg/mL时,阴性T/C=1.0,T、C线显色最明显,阳性试纸条抑制率高达90.0%。因此,选择金标抗体用量为9 μL、包被原4 mg/mL组合。
3.6 标准曲线的建立
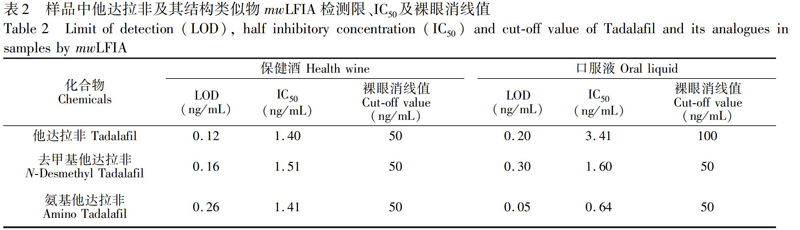
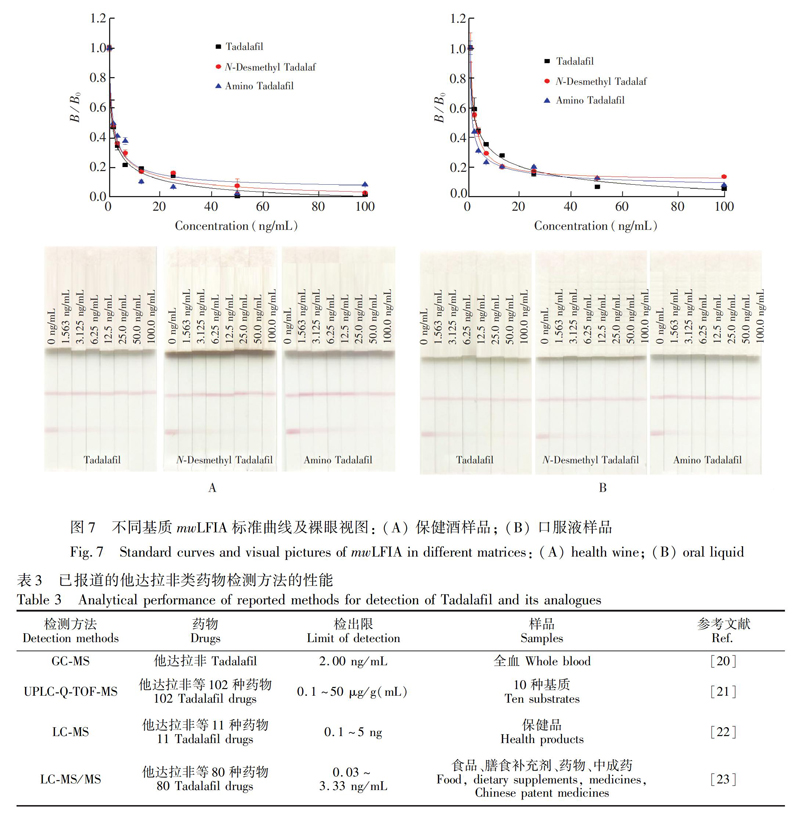
取经HPLCMS/MS确证为阴性的保健酒或口服液样品,按照2.2.10节方法制备阴性样品溶液,将1.0 mg/mL他达拉非类药物溶液稀释,分别配制成100、50 、25、12.5、6.25、3.125、1.563和0 ng/mL他达拉非类药物标准溶液,在最优条件下进行mwLFIA实验,用读数仪读取不同标准品浓度下试纸条T/C值,以B/B0为纵坐标(其中,B0为不加药物时的T/C值,B为药物浓度为x时的T/C值),标准品浓度为横坐标,利用Origin 8.5拟合绘制标准曲线,确定检出限LOD(IC20,B/B0=0.2时所对应的药物浓度)、半抑制浓度(IC50,B/B0=0.5时所对应的药物浓度)及裸眼消线值(T线变开始变为无色对应的药物浓度,即Cutoff值)。结果表明,他达拉非类药物的LOD为0.05~0.30 ng/mL,IC50为0.64~3.41 ng/mL,裸眼消线值在50 ~100 ng/mL之间(图7和表2)。目前,检测他达拉非类药物含量多采用仪器方法(表3),本方法不仅可达到相同水平的检出限,并且具有成本低、操作简便、快速等优势,具有良好的实际应用价值。
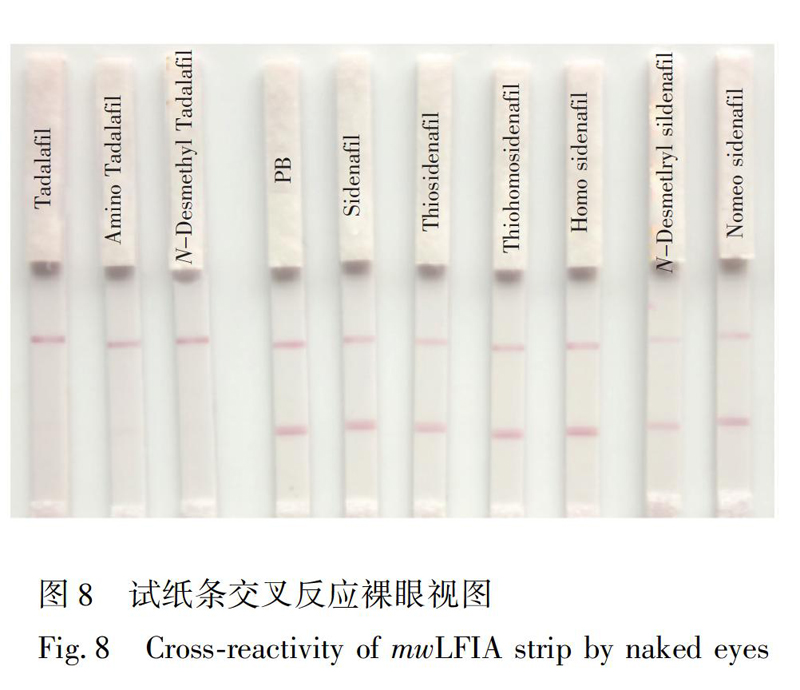
3.7 方法的特异性和稳定性
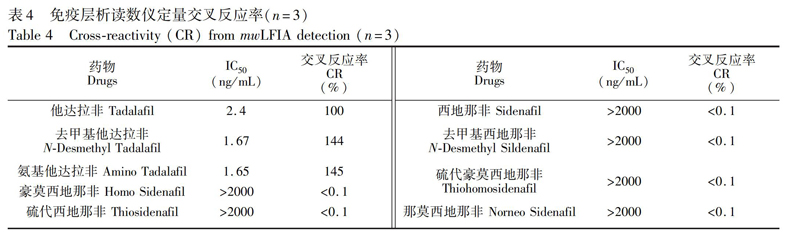
按公式(1)计算交叉反应率(Crossreactivity, CR),评价抗体特异性。
为他达拉非结构功能类似物的IC50。将他达拉非及其类似物配制成200 ng/mL溶液,进行mwLFIA测定,与PB缓冲液对照(图8和表4)。结果表明,本方法可对他达拉非、氨基他达拉非和去甲基他达拉非特异性识别,而與其它结构功能类似物无明显交叉反应,说明此抗体针对他达拉非、氨基他达拉非以及去甲基他达拉非的特异性良好。
将试纸条和金标微孔放入铝箔袋中,37℃干燥保存,分别于第1、3、5、7天取出,采用本方法进行检查,几次检测结果相近,C、T线的颜色变化不大,灵敏度不变,表明本方法稳定性良好。
3.8 实际样品分析
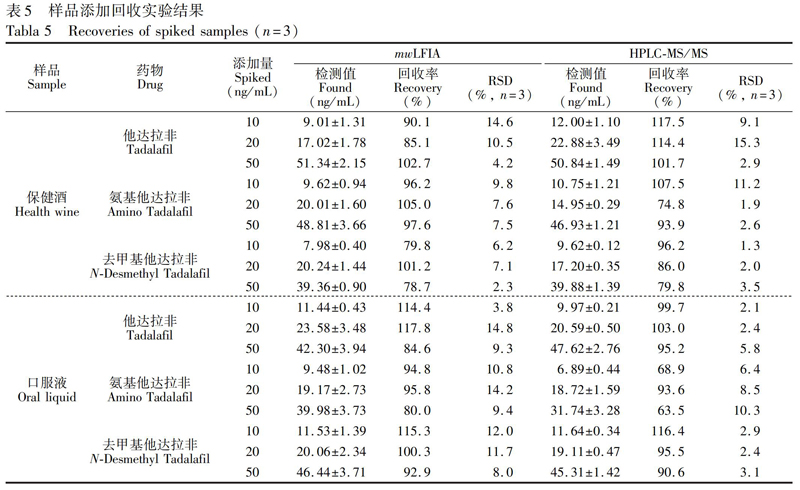
向阴性保健酒和口服液样品中添加他达那非类药物标准品,使终浓度为10、20和50 ng/mL,按照2.2.10节进行样品前处理后,分别采用mwLFIA和HPLCMS/MS方法[24]测定。结果(表5)表明,mwLFIA方法的平均回收率为78.7%~117.8%,RSD <15%,与HPLCMS/MS方法检测结果一致,说明本研究建立的免疫层析检测方法准确可靠,可用于实际样品检测。4 结 论
本研究建立了保健酒和口服液样品中他达拉非、氨基他达拉非和去甲基他达拉非同时检测的mwLFIA方法,检出限为0.05~0.30 ng/mL,裸眼消线值在50~100 ng/mL之间,样品添加回收率为78.7%~117.8%,与HPLCMS/MS法检测结果一致。本方法灵敏、快速、稳定性好,适用于保健酒和口服液基质中他达拉非类药物的现场快速筛查。
References
1 LI Ke, GUO ChangChuan, SHI Feng, ZENG Su, JIANG Wei. Chinese Journal of Pharmaceutical Analysis, 2018, 38(4): 566-574
李 可, 郭常川, 石 峰, 曾 苏, 姜 玮. 药物分析杂志, 2018,? 38(4): 566-574
2 Patel D N, Li L, Ge X W, Kee C L, Low L Y, Koh H L. J. Pharm. Biomed. Anal., 2014,? 87: 176-190
3 LEI Yi, HUANG YanTing, LUO ZhuoYa. Physical Testing and Chemical Analysis(Part B: Chemical Analysis), 2014,? 50(5):? 530-535
雷 毅, 黃艳婷, 罗卓雅.? 理化检验(化学分册),? 2014,? 50 (5) :? 530-535
4 CHEN YuHao, LIANG MoGang, LAN KangHua, GU LiJun. Guangdong Chemical Industry, 2018,? 45(10): 77-79
陈玉浩, 梁茉刚, 蓝康华, 古丽君.? 广东化工,? 2018,? 45(10): 77-79
5 HUANG ZhaoHui, CAI DanDan, CHEN ZhongYi, ZHOU Zheng. Chinese Pharmaceutical Journal, 2015,? 50(4):? 371-374
黄朝辉, 蔡丹丹, 陈仲益, 周 征.? 中国药学杂志,? 2015,? 50(4):? 371-374
6 WANG ZhaoHui, ZHAO HongBing, JIANG XiaoHuang, SHI JiLian. China Pharmacist, 2012,? 15(6): 895
王朝晖, 赵宏冰, 蒋晓煌, 石继连.? 中国药师,? 2012,? 15(6): 895
7 Bojanapu A, Subramaniam A, Munusamy J, Dhanapal K, Chennakesavalu J, Sellappan M, Jayaprakash V. Drug Res., 2015,? 65(2):? 82-85
8 Ma B, Shang X J, Zhang Q, Jing L, Liu Y H, Cao X M, Xu Q Y. J. Pharmaceut. Biomed., 2013,? 77:? 149-157
9 Nikolaou P, Papoutsis I, Athanaselis S, Alevisopoulos G, Khraiwesh A, Pistos C, Spiliopoulou C. J. Pharm. Biomed., 2011,? 56(3):? 577-781
10 Peng J, Liu L Q, Xu C L, Song S S, Kuang H, Cui G. Nano Res., 2017,? 10(1):? 108-120
11 Guo L L, Wu X L, Kuang H, Xu C L. Small, 2018,? 14(6):? 1701782
12 Shen Y D, Wang Y, Zhang S W, Xiao Z L, Sun Y M, Bu X Z, Gu L Q. Chin. Chem. Lett., 2007,? 18(12): 1490-1492
13 SHEN YuDong, DENG LiHua, YANG JinYi, XU ZhenLin, HUA YanTao, WANG Hong, XIAO ZhiLi, LEI HongTao, SUN YuanMing. China Patent, 201510782683.5, 2015
沈玉栋, 邓丽华, 杨金易, 徐振林, 华彦涛, 王 弘, 肖治理, 雷红涛, 孙远明.? 中国专利,? 201510782683.5, 2015
14 LI Ran, LIN ZeJia, YANG JinYi, XU ZhenLin, WANG Hong, LEI HongTao, SUN YuanMing, SHEN YuDong. Chinese J. Anal. Chem., 2018,?? 46(8): 1321-1328
李 然, 林泽佳, 杨金易, 徐振林, 王 弘, 雷红涛, 孙远明, 沈玉栋.? 分析化学,? 2018,? 46(8): 1321-1328
15 YAO ChanYuan, YANG JinYi, XU ZhenLin, WANG Hong, LEI HongTao, SUN YuanMing, TIAN YuanXin, SHEN YuDong.Chinese J. Anal. Chem., 2018,? 46(8):? 1275-1281
姚婵媛, 杨金易, 徐振林, 王 弘, 雷红涛, 孙远明, 田元新, 沈玉栋.? 分析化学,? 2018,? 46(8):? 1275-1281
16 Xu Z L, Shen Y D, Sun Y M, Campbell K, Tian Y X, Zhang S W, Lei H T, Jiang Y M.Talanta, 2013,? 103:? 306-313
17 LIU RuoFei, ZHANG Cui, GE RuYi, CHEN YuQiong, LIU XiaoBo. Chinese Journal of Cellular and Molecular Immunology, 2013,? 29(12):? 1303-1306
劉若飞, 张 萃, 葛如意, 陈玉琼, 刘晓波.? 细胞与分子免疫学杂志,? 2013,? 29(12):? 1303-1306
18 He F, Tian Y X, Xu Z L, Luo L, Yang J Y, Wang H, Sun Y M, Du Q F, Shen Y D. J. Toxicol. Env. Heal. A, 2018,? 81(4): 80-88
19 Zhang G, Wang X, Zhi A, Bao Y, Yang Y, Qu M, Luo J, Li Q, Guo J, Wang Z, Yang J, Xing G, Chai S, Shi T, Liu Q. Food Addit. Contam., 2008,? 25(4):? 413-423
20 Nikolaou P, Papoutsis l, Athanaselis S, Alevisopoulos G, Khraiwesh A, Pistos C, Spiliopoulou C. J. Pharm. Biomed., 2011,? 56(3): 577-581
21 YU Hong, HU Qing, SUN Jian, FENG Rui, ZHANG Su, ZHANG JingXian, MAO XiuHong, JI Shen. Chinese Journal of Chromatography, 2018,? 36(10):? 1005-1017
于 泓, 胡 青, 孙 健, 冯 睿, 张 甦, 张静娴, 毛秀红, 季 申.? 色谱,? 2018,? 36(10):? 1005-1017
22 DONG Yu, JIANG Xin, LIU Jing, WANG ZhenHong. Food Safety and Quality Detection Technolog, 2018,? 9(15): 4055-4060
董 宇, 姜 鑫, 刘 静, 王振红.? 食品安全质量检测学报,? 2018,? 9(15): 4055-4060
23 Lee J H, Park H N, Park O R, Kim N S, Park S K, Kang H. Forensic Sci. Int., 2019,? 298: 10-19
24 BJS 201805. Determination of Sildenafils in Food. State Administration for Market Regulation of the People's Republic of China
BJS 201805. 食品中那非类物质的测定. 中华人民共和国国家市场监督管理总局
- 网络营销风险指标体系的建立原则和评价方法
- 关于企业人力资源成本控制问题的几点思考
- 流程再造对某铁路企业组织架构的影响
- 企业党务工作者队伍建设的方式方法研究
- 中药材生产及其管理规范
- 石油专用管出口营销策略分析
- 石油钢管产品市场营销策略分析
- 试论石油企业内部控制建设中存在问题
- 电网资产全寿命周期标准化管理的信息整合及优化
- 我国企业的内部控制问题研究
- 联邦分权制的价值创造研究
- 论绿色营销与生态经济两者的关系
- 电子商务时代我国网络银行的战略手段
- 制度安排视角下的中国金融结构调整与经济发展
- 公路事业单位会计制度改革的思考
- 企业财务内控管理体系的构建策略
- 浅析部队医院成本核算存在的问题及解决方式
- 探讨两税合并、税收筹划与盈余管理方式的选择
- 高职院校实训资产管理制度实施研究
- 国企财务管理人员能力的提升与培养策略探究
- 应收账款的优化管理
- 财务管理对新会计制度谨慎性原则的应用分析
- 会计师事务所做大做强背景下受惩戒原因探析
- 风险导向审计模式在审计实务中的运用研究
- 山东省中小企业财务管理问题的研究
- removably
- removal
- removals
- remove
- removeable
- removed
- removement
- remover
- remover's
- removers
- removes
- removing
- remultiplication
- remultiplications
- remunerabilities
- remunerability
- remunerable
- remunerably
- remunerate
- remunerated
- remunerates
- remunerating
- remuneration
- remunerationpackage
- remunerations
- 奕劻
- 奕叶重光
- 奕奕
- 奕奕在目
- 奕奕有神的目光
- 奕奕自喜
- 奕山
- 奕棋
- 奕楸
- 奕经
- 奕谱
- 奖
- 奖上
- 奖下
- 奖主
- 奖他人志气,灭自己威风
- 奖任
- 奖优
- 奖优汰劣
- 奖优罚劣
- 奖借
- 奖借过力,受之有愧
- 奖借过隆,实不敢当
- 奖其功劳
- 奖其劳绩