液相色谱—高分辨飞行时间质谱法快速筛查食品中香港规例农药残留
潘孝博+伊雄海+时逸吟+赵善贞+盛永刚+邓晓军+朱坚+丁卓平
摘 要 建立了液相色谱四级杆串联飞行时间质谱法测定食品中249种香港《食物内残余除害剂规例》农药残留的筛查方法。样品经1%甲酸乙腈提取,改进的QuEChERS方法提取净化,Agilent Poroshell 120 ECC18色谱柱(150 mm×3 mm i.d., 2.7 μm)分离,流动相为0.1%甲酸水和甲醇溶液, 梯度洗脱,电喷雾离子源,正模式下侦测,建立了一级精确质量和二级碎片离子质谱数据库,并且对11种典型食品的基质效应进行考察,基质匹配外标法定量。结果表明,在10~500 μg/kg浓度范围内,249种目标化合物线性关系良好(r>0.99),方法定量限为10~100 μg/kg(S/N≥10),在大米、香菇、黄豆、菠菜、西红柿、西兰花、柚子、韭菜、胡萝卜、生菜、黄瓜中3个添加水平的平均回收率范围分别为23.2%~133.2%, 35.6%~137.6%和38.7%~140.2%,相对标准偏差(RSD)在1.3%~19.2%之间(n=6)。方法操作简便,耗时短,灵敏度高,稳定性好,用于日常筛查检测可显著降低检测成本,具有实际应用价值。
关键词 液相色谱四级杆串联飞行时间质谱法; 筛查; 农药残留
20151116收稿; 20160304接受
本文系上海出入境检验检疫课题(Nos. HK0082015,HK0052015)、上海市技术性贸易措施应对专项(Nos. 14TBT006,15TBT004,15TBT005)和上海市技术标准专项(No. 15DZ0503201)资助
Email: yixh@shciq.gov.cn
1 引 言
2014年8月1日开始实施的香港食物中残余除害剂管理制度《食物内残余除害剂规例》(简称《规例》)规管农药数量360种、7083个限量标准,涉及584种(类)食品、农产品[1],远低于《GB 27632014食品安全国家标准食品中农药最大残留限量》有关限量要求标准,许多限量标准内地未制定。中国内地是香港特区食品的主要供应地,占香港进口总量的9成以上,将直接影响内地每年约47亿美元供港食品农产品[2],《规例》的出台对供港食品提出了更高的质量安全要求,同时也对农药残留检测技术提出挑战。然而,目前我国缺乏针对《规例》中农药的高通量检测技术。针对香港《规例》特别是其独有目标物建立筛查检测方法,对于供港食品监督检测部门提供技术支持,以及我国内地尚未在《规例》中规定残留限量的农药提供检测方法的参考,都具有重要意义。
目前,农药残留检测的方法主要有气相色谱法[3]和液相色谱法[4,5],这些方法主要针对单类别目标化合物检测,仪器灵敏度已逐渐不能完全满足国内外对残留物越来越低及不得检出的限量要求,且定性能力较差。近年来,色谱与单级或串联质谱技术的结合,灵敏度和定性能力的逐步提升,气相色谱质谱联用法和液相色谱串联质谱法已成为农药残留痕量分析的主流技术[6~8],通过选择离子监测模式及多反应监测模式实现多种化合物的同时定性定量检测,由于受仪器扫描速度限制,检测通量有限且只适合已知目标物的检测。随着高分辨质谱技术的发展,飞行时间质谱法凭借高通量、高分辨率、谱库检索、不依靠标准品也可分析等能力开始在痕量分析领域得到应用,配合高通量、全质量数据采集以及谱库检索等功能可以快速、准确对大量化合物筛查测定[9]。研究者也在食品农残检测领域进行了探索性的研究[10~12],然而这些研究主要针对单一基质中多类化合物筛查或多种基质中一类化合物筛查,且前处理较为复杂,试剂用量大,检测周期长等特点,不能适应实际工作中遇到的多种复杂基质中跨类别化合物非定向高通量筛查的要求。
本研究建立了应对香港《规例》多组分高通量筛查系统平台,根据目标物性质不同,采用LCQTOF技术可检测249种化合物。 本实验采用改进的QuEChERS法进行前处理,应用LCQTOF技术,实现《规例》中249种目标物在11种典型食品基质中残留量的定性定量检测。本方法将液相四级杆串联飞行时间质谱高分离定性能力与QuEChERS前处理快速灵敏高效等特点充分结合,具有前处理易操作、检测周期短、筛查通量高、定性准确等特点。
2 实验部分
2.1 仪器和试剂
Agilent G6540液相色谱四级杆串联飞行时间质谱仪,配有Dual Agilent Jet Stream Electrospray Ionization (Dual AJS ESI)源; MilliQ超纯水机(美国Millipore公司); PL602L电子天平(瑞士MettlerToledo公司); Allegra X30R Centrifuge离心机; 乙腈、甲醇( HPLC纯,迪马科技有限公司); 乙酸铵、无水MgSO4、NaCl、二水合柠檬酸钠、柠檬酸二钠盐均为分析纯(国药集团化学试剂有限公司); 甲酸(纯度98%,德国Fluka公司); 十八烷基键合硅胶吸附剂(C18)、N丙基乙二胺吸附剂(PSA)、石墨化炭黑(GCB)(上海安谱实验科技股份有限公司); 249种农药标准品: 纯度≥95%(Dr.Ehrenstorfer GmbH)。
2.2 色谱质谱条件
Agilent Poroshell 120 ECC18色谱柱(150 mm×3 mm i.d., 2.7 μm); 柱温40℃; 进样量3 μL; 流动相: A相为0.1%(V/V)甲酸溶液(含5 mmol/L乙酸铵),B相为甲醇溶液。梯度洗脱程序: 0~1.0 min,95% A; 1.0~6.0 min,95%~40% A; 6.0~16.0 min, 40%~0% A; 16.0~20.0 min, 0% A; 20.0~25.0 min,0~95% A。流速: 0.4 mL/min。
离子源: Dual AJS ESI源; 扫描方式: 正离子扫描; 扫描范围m/z 50~1000; 毛细管电压: 4000 V; 鞘气温度: 350℃; 鞘气流速: 11 L/min; 干燥气温度: 280℃; 干燥气流速: 9 L/min; 雾化气压力: 40 psi; 去簇电压: 125 V; 锥孔电压: 65 V; 八级杆射频电压: 750 V。
2.3 样品前处理
2.3.1 提取 准确称取10.0 g新鲜果蔬样品(谷物称取5.0 g样品,加入5 mL水),于50 mL塑料离心管中,加入10 mL 1%甲酸乙腈提取,快速加入4.0 g MgSO4、1.0 g NaCl、1.0 g二水合柠檬酸钠、0.5 g柠檬酸二钠盐,剧烈振荡1 min,静置5 min,再次剧烈振荡1 min,4500 r/min离心10 min。移取上清液6 mL至15 mL离心管。
2.3.2 净化 在上述装有上清液的离心管中加入150.0 mg C18,150.0 mg PSA,900.0 mg MgSO4,15.0 mg GCB粉末,振荡混匀2 min,4500 r/min离心5 min,移取4 mL上清液,加入40 μL含5%甲酸的乙腈,混匀。取0.5 mL上述提取液,乙腈定容至1 mL,过膜后检测。
2.4 数据库的建立
2.4.1 一级精确质量数据库 在优化的色谱质谱条件下进样,对249种化合物先进行一级质谱全扫描,获得目标物的保留时间、母离子、质量偏差,以及离子化形式等信息。通过在PCDL数据库软件中输入每种农药的名称、保留时间、分子式、精确相对分子质量、CAS号,建立了249种化合物的一级精确质量数据库。
2.4.2 二级碎片离子数据库 将目标物精确质量母离子在不同碰撞能量下(10, 20, 30和45 eV)进行测定,采集二级谱图库所需要的子离子信息,通过PCDL软件,建立化合物二级信息谱库,包括母离子、保留时间、不同碰撞能量下二级全扫描谱图。
3 结果与讨论
3.1 色谱柱的优化
为了取得更好的分离效果,实验对比了Agilent ZORBAX RRHD Eclipse Plus C18全多孔填料色谱柱(100 mm×3.0 mm i.d., 1.8 μm)与Agilent Poroshell 120 ECC18表面多孔填料色谱柱(150 mm×3 mm i.d., 2.7 μm)的柱效。结果显示: Poroshell 120 ECC18的柱效为Eclipse Plus C18的90%,但压力却只有其60%。原因可能是由于表面多孔微粒色谱柱填料结构的作用[13]。由于存在实心核,与全多孔颗粒填料相比,溶质具有更短的扩散通道,可有效减少色谱峰展宽,提高柱效。当Poroshell 120 ECC18色谱柱与ZORBAX RRHD Eclipse Plus C18色谱柱在相同柱压时,前者柱效更高,较低的柱压对于色谱柱的使用寿命,以及仪器的日常维护都具有重要作用,所以实验最终选用Poroshell 120 ECC18色谱柱。
3.2 流动相的选择
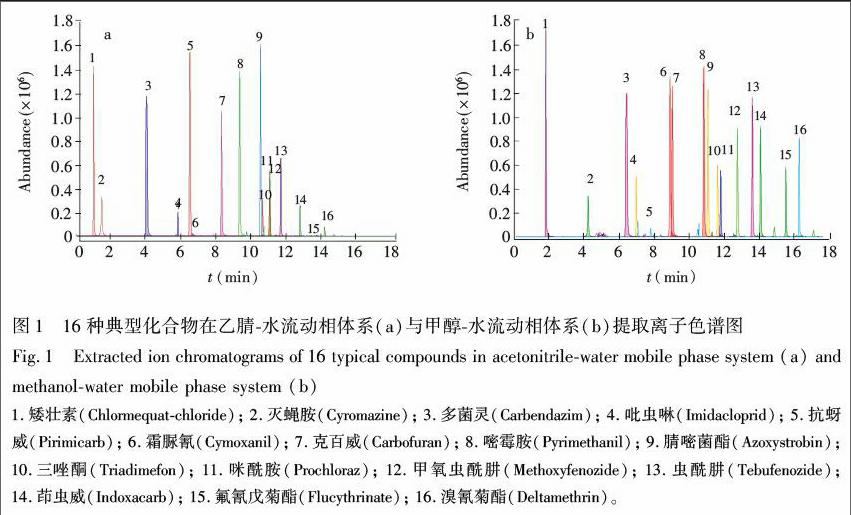
本实验根据极性大小为指标将化合物分类,并选择了16种典型物质作为前处理技术优化的研究对象。弱极性物质选择菊酯类农药溴氰菊酯与氟氰戊菊酯,强极性物质选择季铵盐类农药矮壮素以及三嗪类农药灭蝇胺,中等极性物质分别选择氨基甲酸酯类、烟碱类、三唑类、咪唑类、苯氨基嘧啶类、氰乙酰胺类、甲氧基丙烯酸酯类等12种农药。对以上16种物质分别在乙腈水与甲醇水流动相体系(水相中分别添加0.1%甲酸和5 mmol/L乙酸铵)色谱表现进行对比研究。实验表明,在不同流动相下目标化合物的分离度存在差异,并且质谱响应强弱不同。在乙腈流动相体系,目标物出峰时间在1.0~14.2 min之间, 而在甲醇流动相体系,目标物在1.8~16.2 min之间出峰(图1),物质在甲醇流动相体系中保留能力较强,例如灭蝇胺在乙腈水流动相条件下无保留,而在甲醇水流动相条件下保留情况良好,可能是由于乙腈和甲醇的洗脱能力以及粘度差异导致[14]。在质谱响应强度方面,化合物在甲醇水流动相体系比在乙腈水流动相体系响应值高,这与蓝芳等[15]的研究结果一致,推测原因为乙腈是非质子溶剂,甲醇是质子溶剂,甲醇OH中的H可微弱电离,母离子受生成的加H形式的物质离子促进,故其峰面积提高。因此,本实验采用0.1%甲酸(含5 mmol/L乙酸铵)甲醇作为最优流动相。
3.3 样品前处理方法优化
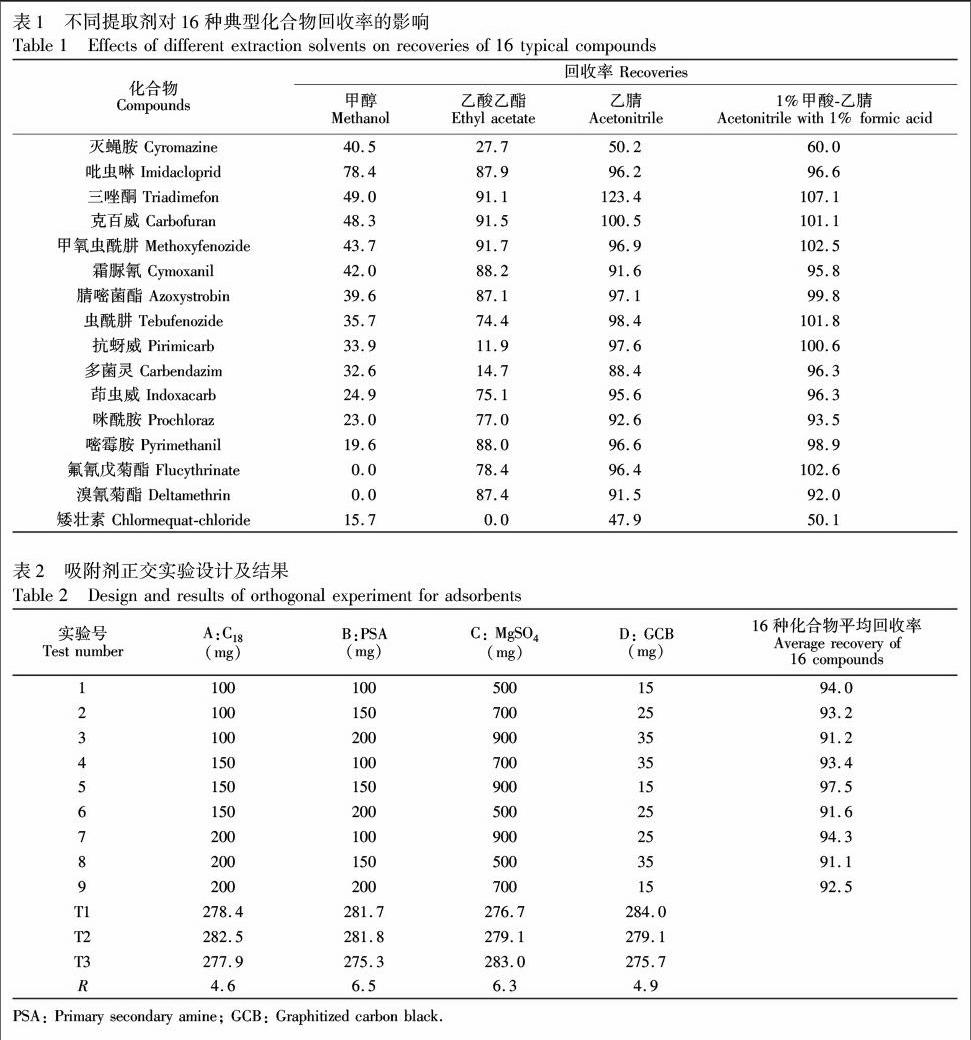
3.3.1 提取溶剂的优化 分别考察了乙酸乙酯、甲醇、乙腈和1%甲酸乙腈对16种典型的化合物的提取效率,标准品用空白基质液配制,以黄瓜基质中加标浓度为10 μg/kg目标物的回收率为考察指标。结果显示(表1),采用甲醇提取时共提物色素较少,回收率范围为0%~49.0%,其中氟氰戊菊酯与溴氰菊酯无法回收。乙酸乙酯提取时,矮壮素无法回收,抗蚜威与多菌灵回收率小于20%。用乙腈提取时,目标物回收率范围在47.9%~123.4%之间。而采用1%甲酸乙腈提取时,考察的化合物回收率均在50.1%~107.1%之间,提取效果最佳,因此本实验选择1%甲酸乙腈作为提取溶剂。
3.3.2 正交实验优化吸附剂 采用正交实验优化QuEChERS净化组合粉末并比较实验因素主效应大小。经初步实验选定C18,PSA,GCB,MgSO4为净化效果的主要影响因素,每种因素选择3个水平,设计L9(34)4因素3水平实验,以黄瓜基质中16种典型化合物平均回收率作为评定依据,通过正交找到最优净化组合。正交实验详情如表2所示。
结果表明,影响回收率的因素主次为PSA>MgSO4>GCB>C18,即PSA对回收率的影响最大,MgSO4次之,C18比较小。考察A, B, C, D 4因素在3个水平上的变化,最佳的净化组合为A2B2C3D1,即150.0 mg C18,150.0 mg PSA,900.0 mg MgSO4,15.0 mg GCB时目标化合物平均回收率最高,净化效果最好,本实验采用该组合作为最优净化方案。
3.4 质谱条件的优化
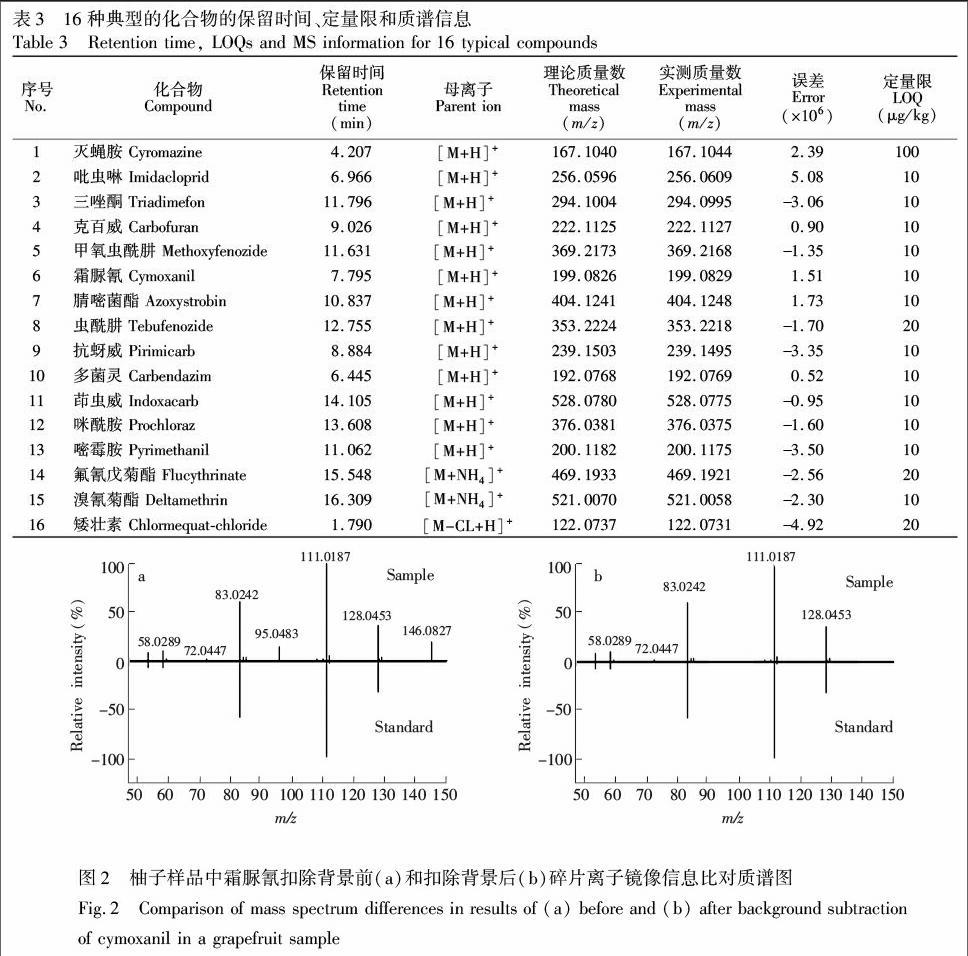
3.4.1 母离子加合形式的选择 离子源为电喷雾双喷源,数据在正模式下采集,有225种化合物母离子加合形式为[M+H]+; 18种化合物加合形式为[M+NH4]+,一些菊酯类物质母离子为[M+NH4]+时响应较好,并且响应值在甲醇流动相中比在乙腈流动相中高(见图1),刘浩等[16]的研究也发现相同现象; 水胺硫磷与溴螨酯监测母离子加合形式为[M+Na]+; 矮壮素加合形式为[M-CL+H]+,王金花等[17]的研究也有相同发现; 敌草快监测母离子加和形式为[M2++H]+,这与李捷等[18]实验结论相同; 有些化合物在离子化过程中会发生中性丢失现象,如百草枯与三环锡,加合形式分别为[M-H2O+H]+及[M-CH3+H]+。因篇幅有限,现将16种典型化合物保留时间、定量限及质谱信息整理如表3所示。
3.4.2 二级质谱最佳碰撞能量优化
对每种物质采集10, 20, 30, 45 eV能量下二级质谱图,根据不同碰撞能量下母离子碎裂程度,选择母离子特征离子明显,碎片离子分布均匀,响应良好的二级质谱图下的碰撞能量为最佳碰撞能量。
3.4.3 二级质谱定性确证 通过对目标物进行一级精确质量质谱分析,会有出现假阳性结果的可能,此时需对疑似化合物进行二级子离子信息确证。确证过程为: 在Targeted MS/MS采集模式下,输入侦测疑似目标物母离子,保留时间及最佳碰撞能量,测定结果用二级质谱库检索,根据欧盟SANCO/12571/2013决议[19],当有两个及两个以上的特征离子匹配,则可确证其为目标物。在确证的过程中为了排除杂质离子对检索的干扰,常需扣除背景。以霜脲氰为例,图2为柚子基质中背景扣除前后镜像比对质谱图。实验发现,在样品背景中含有m/z 95.0483和m/z 146.0827两个干扰离子,从而使碎片离子的比例发生改变, 造成匹配检索结果分值过低,扣除背景后干扰离子被排除,子离子的相对比例正常,提高了匹配分值,达到常规检索确证得分60分的要求,这样可以直接通过谱图检索得分情况对化合物进行确证,而不用进行手动比对子离子匹配排查,大大节约数据处理时间。
3.5 基质效应的评价
液相色谱串联质谱中的基质效应由分析物的共流出组分影响电喷雾接口的离子化效率所致,表现为离子增强或抑制作用[20]。实验考察了11种典型食品的基质效应,在纯试剂和不同基质提取液后添加标样,比较目标物在纯试剂和基质液中的信号峰面积,得到基质响应相对强度(ME)为(基质标样的峰面积/试剂标样的峰面积)×100%[21]。结果表明,在高含水量的果蔬中,如生菜、黄瓜、西红柿、新鲜黄豆中至少有14种物质基质效应在80%~120%之间,基质效应不明显。在大米基质中有11种物质基质效应<80%,表现为基质抑制效应,可能是较低的含水量浓缩了基质共萃物,导致基质效应较显著。除此之外,在高蛋白低脂肪的鲜香菇有9种物质基质效应<80%,表明在高含水量的基质中,蛋白质含量对基质抑制的效果形成有一定影响,这与报道过的基质组分对基质效应的影响结论类似[22]。虫酰肼在大米、菠菜、西兰花、柚子、胡萝卜中基质效应>120%,表现基质增强效应,可能与该物质的性质有关。本实验选用基质匹配溶液校正方法对基质效应补偿,保证检测结果的准确性。
3.6 方法学验证
3.6.1 线性范围和定量限 以各种空白基质标准溶液绘制标准曲线,各浓度点分别为10, 20, 50, 100和500 μg/kg。以峰面积y为纵坐标,浓度x为横坐标,各目标化合物在10~500 μg/kg范围内线性关系良好,相关系数(r)均大于0.99。在基质空白样品中添加标准溶液,以S/N≥10确定方法定量限。结果显示,目标物中有163种物质定量限为10 μg/kg,33种物质定量限为20 μg/kg,36种物质定量限为50 μg/kg,17种物质定量限为100 μg/kg。
3.6.2 回收率及精密度 选用11种食品基质考察本方法的准确性,基质选择涉及成分较复杂的高含水量蔬菜,如菠菜、韭菜等; 也包括低水分、低脂肪、高淀粉谷物类如大米、黄豆; 以及高酸、高含水量柑橘类水果柚子。在上述空白基质中做浓度为10、50和100 μg/kg的添加回收实验,在2.2节条件下测定,每个添加浓度水平做6份平行样品。249种化合物在大米、香菇、黄豆、菠菜、西红柿、西兰花、柚子、韭菜、胡萝卜、生菜、黄瓜中3个添加水平下的平均回收率分别为23.2%~133.2%, 35.6%~137.6%和38.7%~140.2%,相对标准偏差(RSD)在1.3%~19.2%之间(n=6)。
3.7 实际样品测定
实验对某地区534例市售水果蔬菜进行检测,其中383例样品未检出,检出率28.3%。葡萄、草莓、芹菜、青菜等样品的农药检出率较高。检出频率较高的为多菌灵和烯酰吗啉等杀菌剂和杀虫剂。
4 结 论
建立了针对香港《规例》中249种化合物的LCQTOF筛查与确证方法,对提取试剂、流动相组成、净化吸附剂组合、质谱条件进行优化,并对基质效应进行了评价,本实验方法将QuEChERS前处理与高分辨质谱检测结合,可实现11种复杂基质中目标化合物快速准确筛查分析,本方法快速、高效、准确度高,回收率符合国际上痕量分析的规定。
References
1 Hong Kong Department of Justice Bilingual Laws Information System, Pesticide Residues in Food Regulation (CAP132M), http://translate.legislation.gov.hk/gb/www.legislation.gov.hk/blis_pdf.nsf/6799165D2FEE3FA94825755E0033E532/841E356CA9DBCE94482579F30053D610?OpenDocument&bt=0, 2014
2 L Tian. China Food, 2014, (18): 58-59
吕 田. 中国食品, 2014, (18): 58-59
3 Shen Z G, He Z Y, Wang P, Zhou Z Q, Sun M J, Li J D, Liu D H. Anal. Chim. Acta, 2013, 793: 37-43
4 Li N, Chen J, Shi Y P. Talanta, 2015, 141: 212-219
5 RAO Zhu, LI Song, HE Miao, SU Jin. Chinese J. Anal. Chem., 2007, 35(7): 954-958
饶 竹, 李 松, 何 淼, 苏 劲. 分析化学, 2007, 35(7): 954-958
6 Mujawar S, Utture S C, Fonseca E, Matarrita J, Banerjee K. Food Chem., 2014, 150(5): 175-181
7 Kasiotis K M, Anagnostopoulos C, Anastasiadou P, Machera K. Sci. Total Environ., 2014, 485486(7): 633-642
8 YAN Rui, SHAO MingYuan, J FuLong, SONG DaQian, ZHANG HanQi, Y AiMin. Chinese J. Anal Chem., 2013, 41(2): 315-316
闫 蕊, 邵明媛, 鞠福龙, 宋大千, 张寒琦, 于爱民. 分析化学, 2013, 41(2): 315-316
9 Meng Z, Shi Z H, Liang S X, Dong X F, Lv Y K, Sun H W. Food Control, 2015, 55: 158-165
10 Sivaperumal P, Anand P, Riddhi L. Food Chem., 2015, 168: 356-365
11 YU Lu, SONG Wei, L YaNing, ZHAO MuYu, ZHOU FangFang, HU YanYun, ZHENG Ping. Chinese Journal of Chromatography, 2015, 33(6): 597-612
余 璐, 宋 伟, 吕亚宁, 赵暮雨, 周芳芳, 胡艳云, 郑 平. 色谱, 2015, 33(6): 597-612
12 ZHAO ZhiYuan, SHI ZhiHong, KANG Jian, PENG Xing, CAO XinYue, FAN ChunLin, PANG GuoFang, L MeiLing. Chinese Journal of Chromatography, 2013, 31(4): 372-379
赵志远, 石志红, 康健, 彭 兴, 曹新悦, 范春林, 庞国芳, 吕美玲. 色谱, 2013, 31(4): 372-379
13 ZHU Jian. Journal of Inspection and Quarantine, 2012, 22(5): 1-7
朱 坚. 检验检疫学刊, 2012, 22(5): 1-7
14 WU Cheng, ZHAO ZhiQiang. Chinese Journal of Analysis Laboratory, 2015, 34(6): 711-716
吴 成, 赵志强. 分析试验室, 2015, 34(6): 711-716
15 LAN Fang, ZHANG Yi, YUE ZhenFeng, LIN ShanShan, WU WeiDong, WU FengQi, ZHAO FengJuan, ZHANG Feng, CHENG Cheng. Journal of Instrumental Analysis, 2012, 12(31): 1471-1478
蓝 芳, 张 毅, 岳振峰, 林珊珊, 吴卫东, 吴凤琪, 赵凤娟, 张 峰, 程 程. 分析测试学报, 2012, 12(31): 1471-1478
16 LIU Hao, GAN ZhiYong. Environmental Science and Management, 2014, 39(10): 134-143
刘 浩, 甘志永. 环境科学与管理, 2014, 39(10): 134-143
17 WANG JinHua, LU XiaoYu, HHUANG Mei, WU Zhuan, MA GuiPing, XU ChaoYi. Chinese J. Anal Chem., 2007, 35(10): 1509-1512
王金花, 卢晓宇, 黄 梅, 吴 瑑, 马贵平, 徐超一. 分析化学, 2007, 35(10): 1509-1512
18 LI Jie, YANG Fang, LU ShengYu, LIU ZhengCai, WANG Yan, LAN JinChang, JIANG JinBin, CHEN YanGui. Chinese Journal of Analysis Laboratory, 2014, 33(5): 537-541
李 捷, 杨 方, 卢声宇, 刘正才, 王 彦, 蓝锦昌, 江锦彬, 陈言贵. 分析试验室, 2014, 33(5): 537-541
19 Guidance Document on Analytical Quality Control and Validation Procedures for Pesticide Residues Analysis in Food and Feed. European Commission Health & Consumer Protection DirectorateGeneral, SANCO/12571/2013
食品及饲料品农药残留分析质量控制和验证程序指导文件. 欧洲委员会健康和消费者保护总司. SANCO/12571/2013
20 Eeckhaut A V, Lanckmans K, Sarre S, Smolders I, Michotte Y. J. Chromatogr. B., 2009, 877(23): 2198-2207
21 Periata A, Kohlera I, Thomasb A, Nicolic R, Boccarda J, Veutheya J L, Schapplera J, Guillarme D. J. Chromatogr. A., DOI:10.1016/j.chroma. 2015. 09.035
22 Godula M, Hajlov J, Alterov K. J. High Resolution Chromatogr., 1999, 22(7): 395-402
Abstract A method for screening of 249 residue compounds of pesticide in foods from the Hongkong′s regulation was developed by liquid chromatography coupled with quadrupole timeofflight mass spectrometry (LCQTOF/MS). The target analytes were dissolved in acetonitrile with 1% formic acid, then extracted and purified by using a modified QuEChERS method. The chromatographic separation was performed on an Agilent Poroshell 120 ECC18 column (150 mm×3 mm i.d., 2.7 μm) with gradient elution using 0.1% (V/V) formic acid and methanol as mobile phase. The target compounds were monitored under positive ionization mode with ESI source. The matrix effects in 11 kinds of typical foods were considered and the quantification was carried out by matrixmatched with external standard method. Two databases of accurate mass and fragment ions were created. The results demonstrated that the linear range was from 10 μg/kg to 500 μg/kg with good correlation coefficients (r>0.90). The method quantitation limits were in the range of 10-100 μg/kg (S/N≥10). For rices, mushrooms, soybeans, spinaches, tomatoes, broccolies, grapefruits, chives, carrots, lettuces and cucumbers, the average recoveries at three spiked levels were in the range of 23.2%-133.2%, 35.6%-137.6%, and 38.7%-140.2%, and the relative standard deviations (RSDs) were 1.3%-19.2% (n=6). The method is simple, timesaving with high sensitivity and good reproducibility, as well as having practical application for its low test cost.
Keywords Liquid chromatography coupled with quadrupole timeofflight mass spectrometry; Pesticide; Screening; Pesticide residue
摘 要 建立了液相色谱四级杆串联飞行时间质谱法测定食品中249种香港《食物内残余除害剂规例》农药残留的筛查方法。样品经1%甲酸乙腈提取,改进的QuEChERS方法提取净化,Agilent Poroshell 120 ECC18色谱柱(150 mm×3 mm i.d., 2.7 μm)分离,流动相为0.1%甲酸水和甲醇溶液, 梯度洗脱,电喷雾离子源,正模式下侦测,建立了一级精确质量和二级碎片离子质谱数据库,并且对11种典型食品的基质效应进行考察,基质匹配外标法定量。结果表明,在10~500 μg/kg浓度范围内,249种目标化合物线性关系良好(r>0.99),方法定量限为10~100 μg/kg(S/N≥10),在大米、香菇、黄豆、菠菜、西红柿、西兰花、柚子、韭菜、胡萝卜、生菜、黄瓜中3个添加水平的平均回收率范围分别为23.2%~133.2%, 35.6%~137.6%和38.7%~140.2%,相对标准偏差(RSD)在1.3%~19.2%之间(n=6)。方法操作简便,耗时短,灵敏度高,稳定性好,用于日常筛查检测可显著降低检测成本,具有实际应用价值。
关键词 液相色谱四级杆串联飞行时间质谱法; 筛查; 农药残留
20151116收稿; 20160304接受
本文系上海出入境检验检疫课题(Nos. HK0082015,HK0052015)、上海市技术性贸易措施应对专项(Nos. 14TBT006,15TBT004,15TBT005)和上海市技术标准专项(No. 15DZ0503201)资助
Email: yixh@shciq.gov.cn
1 引 言
2014年8月1日开始实施的香港食物中残余除害剂管理制度《食物内残余除害剂规例》(简称《规例》)规管农药数量360种、7083个限量标准,涉及584种(类)食品、农产品[1],远低于《GB 27632014食品安全国家标准食品中农药最大残留限量》有关限量要求标准,许多限量标准内地未制定。中国内地是香港特区食品的主要供应地,占香港进口总量的9成以上,将直接影响内地每年约47亿美元供港食品农产品[2],《规例》的出台对供港食品提出了更高的质量安全要求,同时也对农药残留检测技术提出挑战。然而,目前我国缺乏针对《规例》中农药的高通量检测技术。针对香港《规例》特别是其独有目标物建立筛查检测方法,对于供港食品监督检测部门提供技术支持,以及我国内地尚未在《规例》中规定残留限量的农药提供检测方法的参考,都具有重要意义。
目前,农药残留检测的方法主要有气相色谱法[3]和液相色谱法[4,5],这些方法主要针对单类别目标化合物检测,仪器灵敏度已逐渐不能完全满足国内外对残留物越来越低及不得检出的限量要求,且定性能力较差。近年来,色谱与单级或串联质谱技术的结合,灵敏度和定性能力的逐步提升,气相色谱质谱联用法和液相色谱串联质谱法已成为农药残留痕量分析的主流技术[6~8],通过选择离子监测模式及多反应监测模式实现多种化合物的同时定性定量检测,由于受仪器扫描速度限制,检测通量有限且只适合已知目标物的检测。随着高分辨质谱技术的发展,飞行时间质谱法凭借高通量、高分辨率、谱库检索、不依靠标准品也可分析等能力开始在痕量分析领域得到应用,配合高通量、全质量数据采集以及谱库检索等功能可以快速、准确对大量化合物筛查测定[9]。研究者也在食品农残检测领域进行了探索性的研究[10~12],然而这些研究主要针对单一基质中多类化合物筛查或多种基质中一类化合物筛查,且前处理较为复杂,试剂用量大,检测周期长等特点,不能适应实际工作中遇到的多种复杂基质中跨类别化合物非定向高通量筛查的要求。
本研究建立了应对香港《规例》多组分高通量筛查系统平台,根据目标物性质不同,采用LCQTOF技术可检测249种化合物。 本实验采用改进的QuEChERS法进行前处理,应用LCQTOF技术,实现《规例》中249种目标物在11种典型食品基质中残留量的定性定量检测。本方法将液相四级杆串联飞行时间质谱高分离定性能力与QuEChERS前处理快速灵敏高效等特点充分结合,具有前处理易操作、检测周期短、筛查通量高、定性准确等特点。
2 实验部分
2.1 仪器和试剂
Agilent G6540液相色谱四级杆串联飞行时间质谱仪,配有Dual Agilent Jet Stream Electrospray Ionization (Dual AJS ESI)源; MilliQ超纯水机(美国Millipore公司); PL602L电子天平(瑞士MettlerToledo公司); Allegra X30R Centrifuge离心机; 乙腈、甲醇( HPLC纯,迪马科技有限公司); 乙酸铵、无水MgSO4、NaCl、二水合柠檬酸钠、柠檬酸二钠盐均为分析纯(国药集团化学试剂有限公司); 甲酸(纯度98%,德国Fluka公司); 十八烷基键合硅胶吸附剂(C18)、N丙基乙二胺吸附剂(PSA)、石墨化炭黑(GCB)(上海安谱实验科技股份有限公司); 249种农药标准品: 纯度≥95%(Dr.Ehrenstorfer GmbH)。
2.2 色谱质谱条件
Agilent Poroshell 120 ECC18色谱柱(150 mm×3 mm i.d., 2.7 μm); 柱温40℃; 进样量3 μL; 流动相: A相为0.1%(V/V)甲酸溶液(含5 mmol/L乙酸铵),B相为甲醇溶液。梯度洗脱程序: 0~1.0 min,95% A; 1.0~6.0 min,95%~40% A; 6.0~16.0 min, 40%~0% A; 16.0~20.0 min, 0% A; 20.0~25.0 min,0~95% A。流速: 0.4 mL/min。
离子源: Dual AJS ESI源; 扫描方式: 正离子扫描; 扫描范围m/z 50~1000; 毛细管电压: 4000 V; 鞘气温度: 350℃; 鞘气流速: 11 L/min; 干燥气温度: 280℃; 干燥气流速: 9 L/min; 雾化气压力: 40 psi; 去簇电压: 125 V; 锥孔电压: 65 V; 八级杆射频电压: 750 V。
2.3 样品前处理
2.3.1 提取 准确称取10.0 g新鲜果蔬样品(谷物称取5.0 g样品,加入5 mL水),于50 mL塑料离心管中,加入10 mL 1%甲酸乙腈提取,快速加入4.0 g MgSO4、1.0 g NaCl、1.0 g二水合柠檬酸钠、0.5 g柠檬酸二钠盐,剧烈振荡1 min,静置5 min,再次剧烈振荡1 min,4500 r/min离心10 min。移取上清液6 mL至15 mL离心管。
2.3.2 净化 在上述装有上清液的离心管中加入150.0 mg C18,150.0 mg PSA,900.0 mg MgSO4,15.0 mg GCB粉末,振荡混匀2 min,4500 r/min离心5 min,移取4 mL上清液,加入40 μL含5%甲酸的乙腈,混匀。取0.5 mL上述提取液,乙腈定容至1 mL,过膜后检测。
2.4 数据库的建立
2.4.1 一级精确质量数据库 在优化的色谱质谱条件下进样,对249种化合物先进行一级质谱全扫描,获得目标物的保留时间、母离子、质量偏差,以及离子化形式等信息。通过在PCDL数据库软件中输入每种农药的名称、保留时间、分子式、精确相对分子质量、CAS号,建立了249种化合物的一级精确质量数据库。
2.4.2 二级碎片离子数据库 将目标物精确质量母离子在不同碰撞能量下(10, 20, 30和45 eV)进行测定,采集二级谱图库所需要的子离子信息,通过PCDL软件,建立化合物二级信息谱库,包括母离子、保留时间、不同碰撞能量下二级全扫描谱图。
3 结果与讨论
3.1 色谱柱的优化
为了取得更好的分离效果,实验对比了Agilent ZORBAX RRHD Eclipse Plus C18全多孔填料色谱柱(100 mm×3.0 mm i.d., 1.8 μm)与Agilent Poroshell 120 ECC18表面多孔填料色谱柱(150 mm×3 mm i.d., 2.7 μm)的柱效。结果显示: Poroshell 120 ECC18的柱效为Eclipse Plus C18的90%,但压力却只有其60%。原因可能是由于表面多孔微粒色谱柱填料结构的作用[13]。由于存在实心核,与全多孔颗粒填料相比,溶质具有更短的扩散通道,可有效减少色谱峰展宽,提高柱效。当Poroshell 120 ECC18色谱柱与ZORBAX RRHD Eclipse Plus C18色谱柱在相同柱压时,前者柱效更高,较低的柱压对于色谱柱的使用寿命,以及仪器的日常维护都具有重要作用,所以实验最终选用Poroshell 120 ECC18色谱柱。
3.2 流动相的选择
本实验根据极性大小为指标将化合物分类,并选择了16种典型物质作为前处理技术优化的研究对象。弱极性物质选择菊酯类农药溴氰菊酯与氟氰戊菊酯,强极性物质选择季铵盐类农药矮壮素以及三嗪类农药灭蝇胺,中等极性物质分别选择氨基甲酸酯类、烟碱类、三唑类、咪唑类、苯氨基嘧啶类、氰乙酰胺类、甲氧基丙烯酸酯类等12种农药。对以上16种物质分别在乙腈水与甲醇水流动相体系(水相中分别添加0.1%甲酸和5 mmol/L乙酸铵)色谱表现进行对比研究。实验表明,在不同流动相下目标化合物的分离度存在差异,并且质谱响应强弱不同。在乙腈流动相体系,目标物出峰时间在1.0~14.2 min之间, 而在甲醇流动相体系,目标物在1.8~16.2 min之间出峰(图1),物质在甲醇流动相体系中保留能力较强,例如灭蝇胺在乙腈水流动相条件下无保留,而在甲醇水流动相条件下保留情况良好,可能是由于乙腈和甲醇的洗脱能力以及粘度差异导致[14]。在质谱响应强度方面,化合物在甲醇水流动相体系比在乙腈水流动相体系响应值高,这与蓝芳等[15]的研究结果一致,推测原因为乙腈是非质子溶剂,甲醇是质子溶剂,甲醇OH中的H可微弱电离,母离子受生成的加H形式的物质离子促进,故其峰面积提高。因此,本实验采用0.1%甲酸(含5 mmol/L乙酸铵)甲醇作为最优流动相。
3.3 样品前处理方法优化
3.3.1 提取溶剂的优化 分别考察了乙酸乙酯、甲醇、乙腈和1%甲酸乙腈对16种典型的化合物的提取效率,标准品用空白基质液配制,以黄瓜基质中加标浓度为10 μg/kg目标物的回收率为考察指标。结果显示(表1),采用甲醇提取时共提物色素较少,回收率范围为0%~49.0%,其中氟氰戊菊酯与溴氰菊酯无法回收。乙酸乙酯提取时,矮壮素无法回收,抗蚜威与多菌灵回收率小于20%。用乙腈提取时,目标物回收率范围在47.9%~123.4%之间。而采用1%甲酸乙腈提取时,考察的化合物回收率均在50.1%~107.1%之间,提取效果最佳,因此本实验选择1%甲酸乙腈作为提取溶剂。
3.3.2 正交实验优化吸附剂 采用正交实验优化QuEChERS净化组合粉末并比较实验因素主效应大小。经初步实验选定C18,PSA,GCB,MgSO4为净化效果的主要影响因素,每种因素选择3个水平,设计L9(34)4因素3水平实验,以黄瓜基质中16种典型化合物平均回收率作为评定依据,通过正交找到最优净化组合。正交实验详情如表2所示。
结果表明,影响回收率的因素主次为PSA>MgSO4>GCB>C18,即PSA对回收率的影响最大,MgSO4次之,C18比较小。考察A, B, C, D 4因素在3个水平上的变化,最佳的净化组合为A2B2C3D1,即150.0 mg C18,150.0 mg PSA,900.0 mg MgSO4,15.0 mg GCB时目标化合物平均回收率最高,净化效果最好,本实验采用该组合作为最优净化方案。
3.4 质谱条件的优化
3.4.1 母离子加合形式的选择 离子源为电喷雾双喷源,数据在正模式下采集,有225种化合物母离子加合形式为[M+H]+; 18种化合物加合形式为[M+NH4]+,一些菊酯类物质母离子为[M+NH4]+时响应较好,并且响应值在甲醇流动相中比在乙腈流动相中高(见图1),刘浩等[16]的研究也发现相同现象; 水胺硫磷与溴螨酯监测母离子加合形式为[M+Na]+; 矮壮素加合形式为[M-CL+H]+,王金花等[17]的研究也有相同发现; 敌草快监测母离子加和形式为[M2++H]+,这与李捷等[18]实验结论相同; 有些化合物在离子化过程中会发生中性丢失现象,如百草枯与三环锡,加合形式分别为[M-H2O+H]+及[M-CH3+H]+。因篇幅有限,现将16种典型化合物保留时间、定量限及质谱信息整理如表3所示。
3.4.2 二级质谱最佳碰撞能量优化
对每种物质采集10, 20, 30, 45 eV能量下二级质谱图,根据不同碰撞能量下母离子碎裂程度,选择母离子特征离子明显,碎片离子分布均匀,响应良好的二级质谱图下的碰撞能量为最佳碰撞能量。
3.4.3 二级质谱定性确证 通过对目标物进行一级精确质量质谱分析,会有出现假阳性结果的可能,此时需对疑似化合物进行二级子离子信息确证。确证过程为: 在Targeted MS/MS采集模式下,输入侦测疑似目标物母离子,保留时间及最佳碰撞能量,测定结果用二级质谱库检索,根据欧盟SANCO/12571/2013决议[19],当有两个及两个以上的特征离子匹配,则可确证其为目标物。在确证的过程中为了排除杂质离子对检索的干扰,常需扣除背景。以霜脲氰为例,图2为柚子基质中背景扣除前后镜像比对质谱图。实验发现,在样品背景中含有m/z 95.0483和m/z 146.0827两个干扰离子,从而使碎片离子的比例发生改变, 造成匹配检索结果分值过低,扣除背景后干扰离子被排除,子离子的相对比例正常,提高了匹配分值,达到常规检索确证得分60分的要求,这样可以直接通过谱图检索得分情况对化合物进行确证,而不用进行手动比对子离子匹配排查,大大节约数据处理时间。
3.5 基质效应的评价
液相色谱串联质谱中的基质效应由分析物的共流出组分影响电喷雾接口的离子化效率所致,表现为离子增强或抑制作用[20]。实验考察了11种典型食品的基质效应,在纯试剂和不同基质提取液后添加标样,比较目标物在纯试剂和基质液中的信号峰面积,得到基质响应相对强度(ME)为(基质标样的峰面积/试剂标样的峰面积)×100%[21]。结果表明,在高含水量的果蔬中,如生菜、黄瓜、西红柿、新鲜黄豆中至少有14种物质基质效应在80%~120%之间,基质效应不明显。在大米基质中有11种物质基质效应<80%,表现为基质抑制效应,可能是较低的含水量浓缩了基质共萃物,导致基质效应较显著。除此之外,在高蛋白低脂肪的鲜香菇有9种物质基质效应<80%,表明在高含水量的基质中,蛋白质含量对基质抑制的效果形成有一定影响,这与报道过的基质组分对基质效应的影响结论类似[22]。虫酰肼在大米、菠菜、西兰花、柚子、胡萝卜中基质效应>120%,表现基质增强效应,可能与该物质的性质有关。本实验选用基质匹配溶液校正方法对基质效应补偿,保证检测结果的准确性。
3.6 方法学验证
3.6.1 线性范围和定量限 以各种空白基质标准溶液绘制标准曲线,各浓度点分别为10, 20, 50, 100和500 μg/kg。以峰面积y为纵坐标,浓度x为横坐标,各目标化合物在10~500 μg/kg范围内线性关系良好,相关系数(r)均大于0.99。在基质空白样品中添加标准溶液,以S/N≥10确定方法定量限。结果显示,目标物中有163种物质定量限为10 μg/kg,33种物质定量限为20 μg/kg,36种物质定量限为50 μg/kg,17种物质定量限为100 μg/kg。
3.6.2 回收率及精密度 选用11种食品基质考察本方法的准确性,基质选择涉及成分较复杂的高含水量蔬菜,如菠菜、韭菜等; 也包括低水分、低脂肪、高淀粉谷物类如大米、黄豆; 以及高酸、高含水量柑橘类水果柚子。在上述空白基质中做浓度为10、50和100 μg/kg的添加回收实验,在2.2节条件下测定,每个添加浓度水平做6份平行样品。249种化合物在大米、香菇、黄豆、菠菜、西红柿、西兰花、柚子、韭菜、胡萝卜、生菜、黄瓜中3个添加水平下的平均回收率分别为23.2%~133.2%, 35.6%~137.6%和38.7%~140.2%,相对标准偏差(RSD)在1.3%~19.2%之间(n=6)。
3.7 实际样品测定
实验对某地区534例市售水果蔬菜进行检测,其中383例样品未检出,检出率28.3%。葡萄、草莓、芹菜、青菜等样品的农药检出率较高。检出频率较高的为多菌灵和烯酰吗啉等杀菌剂和杀虫剂。
4 结 论
建立了针对香港《规例》中249种化合物的LCQTOF筛查与确证方法,对提取试剂、流动相组成、净化吸附剂组合、质谱条件进行优化,并对基质效应进行了评价,本实验方法将QuEChERS前处理与高分辨质谱检测结合,可实现11种复杂基质中目标化合物快速准确筛查分析,本方法快速、高效、准确度高,回收率符合国际上痕量分析的规定。
References
1 Hong Kong Department of Justice Bilingual Laws Information System, Pesticide Residues in Food Regulation (CAP132M), http://translate.legislation.gov.hk/gb/www.legislation.gov.hk/blis_pdf.nsf/6799165D2FEE3FA94825755E0033E532/841E356CA9DBCE94482579F30053D610?OpenDocument&bt=0, 2014
2 L Tian. China Food, 2014, (18): 58-59
吕 田. 中国食品, 2014, (18): 58-59
3 Shen Z G, He Z Y, Wang P, Zhou Z Q, Sun M J, Li J D, Liu D H. Anal. Chim. Acta, 2013, 793: 37-43
4 Li N, Chen J, Shi Y P. Talanta, 2015, 141: 212-219
5 RAO Zhu, LI Song, HE Miao, SU Jin. Chinese J. Anal. Chem., 2007, 35(7): 954-958
饶 竹, 李 松, 何 淼, 苏 劲. 分析化学, 2007, 35(7): 954-958
6 Mujawar S, Utture S C, Fonseca E, Matarrita J, Banerjee K. Food Chem., 2014, 150(5): 175-181
7 Kasiotis K M, Anagnostopoulos C, Anastasiadou P, Machera K. Sci. Total Environ., 2014, 485486(7): 633-642
8 YAN Rui, SHAO MingYuan, J FuLong, SONG DaQian, ZHANG HanQi, Y AiMin. Chinese J. Anal Chem., 2013, 41(2): 315-316
闫 蕊, 邵明媛, 鞠福龙, 宋大千, 张寒琦, 于爱民. 分析化学, 2013, 41(2): 315-316
9 Meng Z, Shi Z H, Liang S X, Dong X F, Lv Y K, Sun H W. Food Control, 2015, 55: 158-165
10 Sivaperumal P, Anand P, Riddhi L. Food Chem., 2015, 168: 356-365
11 YU Lu, SONG Wei, L YaNing, ZHAO MuYu, ZHOU FangFang, HU YanYun, ZHENG Ping. Chinese Journal of Chromatography, 2015, 33(6): 597-612
余 璐, 宋 伟, 吕亚宁, 赵暮雨, 周芳芳, 胡艳云, 郑 平. 色谱, 2015, 33(6): 597-612
12 ZHAO ZhiYuan, SHI ZhiHong, KANG Jian, PENG Xing, CAO XinYue, FAN ChunLin, PANG GuoFang, L MeiLing. Chinese Journal of Chromatography, 2013, 31(4): 372-379
赵志远, 石志红, 康健, 彭 兴, 曹新悦, 范春林, 庞国芳, 吕美玲. 色谱, 2013, 31(4): 372-379
13 ZHU Jian. Journal of Inspection and Quarantine, 2012, 22(5): 1-7
朱 坚. 检验检疫学刊, 2012, 22(5): 1-7
14 WU Cheng, ZHAO ZhiQiang. Chinese Journal of Analysis Laboratory, 2015, 34(6): 711-716
吴 成, 赵志强. 分析试验室, 2015, 34(6): 711-716
15 LAN Fang, ZHANG Yi, YUE ZhenFeng, LIN ShanShan, WU WeiDong, WU FengQi, ZHAO FengJuan, ZHANG Feng, CHENG Cheng. Journal of Instrumental Analysis, 2012, 12(31): 1471-1478
蓝 芳, 张 毅, 岳振峰, 林珊珊, 吴卫东, 吴凤琪, 赵凤娟, 张 峰, 程 程. 分析测试学报, 2012, 12(31): 1471-1478
16 LIU Hao, GAN ZhiYong. Environmental Science and Management, 2014, 39(10): 134-143
刘 浩, 甘志永. 环境科学与管理, 2014, 39(10): 134-143
17 WANG JinHua, LU XiaoYu, HHUANG Mei, WU Zhuan, MA GuiPing, XU ChaoYi. Chinese J. Anal Chem., 2007, 35(10): 1509-1512
王金花, 卢晓宇, 黄 梅, 吴 瑑, 马贵平, 徐超一. 分析化学, 2007, 35(10): 1509-1512
18 LI Jie, YANG Fang, LU ShengYu, LIU ZhengCai, WANG Yan, LAN JinChang, JIANG JinBin, CHEN YanGui. Chinese Journal of Analysis Laboratory, 2014, 33(5): 537-541
李 捷, 杨 方, 卢声宇, 刘正才, 王 彦, 蓝锦昌, 江锦彬, 陈言贵. 分析试验室, 2014, 33(5): 537-541
19 Guidance Document on Analytical Quality Control and Validation Procedures for Pesticide Residues Analysis in Food and Feed. European Commission Health & Consumer Protection DirectorateGeneral, SANCO/12571/2013
食品及饲料品农药残留分析质量控制和验证程序指导文件. 欧洲委员会健康和消费者保护总司. SANCO/12571/2013
20 Eeckhaut A V, Lanckmans K, Sarre S, Smolders I, Michotte Y. J. Chromatogr. B., 2009, 877(23): 2198-2207
21 Periata A, Kohlera I, Thomasb A, Nicolic R, Boccarda J, Veutheya J L, Schapplera J, Guillarme D. J. Chromatogr. A., DOI:10.1016/j.chroma. 2015. 09.035
22 Godula M, Hajlov J, Alterov K. J. High Resolution Chromatogr., 1999, 22(7): 395-402
Abstract A method for screening of 249 residue compounds of pesticide in foods from the Hongkong′s regulation was developed by liquid chromatography coupled with quadrupole timeofflight mass spectrometry (LCQTOF/MS). The target analytes were dissolved in acetonitrile with 1% formic acid, then extracted and purified by using a modified QuEChERS method. The chromatographic separation was performed on an Agilent Poroshell 120 ECC18 column (150 mm×3 mm i.d., 2.7 μm) with gradient elution using 0.1% (V/V) formic acid and methanol as mobile phase. The target compounds were monitored under positive ionization mode with ESI source. The matrix effects in 11 kinds of typical foods were considered and the quantification was carried out by matrixmatched with external standard method. Two databases of accurate mass and fragment ions were created. The results demonstrated that the linear range was from 10 μg/kg to 500 μg/kg with good correlation coefficients (r>0.90). The method quantitation limits were in the range of 10-100 μg/kg (S/N≥10). For rices, mushrooms, soybeans, spinaches, tomatoes, broccolies, grapefruits, chives, carrots, lettuces and cucumbers, the average recoveries at three spiked levels were in the range of 23.2%-133.2%, 35.6%-137.6%, and 38.7%-140.2%, and the relative standard deviations (RSDs) were 1.3%-19.2% (n=6). The method is simple, timesaving with high sensitivity and good reproducibility, as well as having practical application for its low test cost.
Keywords Liquid chromatography coupled with quadrupole timeofflight mass spectrometry; Pesticide; Screening; Pesticide residue
