

摘要 研究了193 nm ArF准分子激光剥蚀系统高空间分辨率下的仪器检出限、ICP质量负载元素分馏、剥蚀深度/束斑直径元素分馏以及基体效应,并在10 μm束斑直径下分析了GSD-1G、StHs6/80-G和NIST612中的微量元素。结果表明,仪器检出限随束斑直径的减小而升高,当束斑直径降低至7 μm时,部分微量元素的仪器检出限为1~10 μg/g。ICP质量负载元素分馏指数与元素第一电离能呈正相关和元素氧化物熔点呈负相关。当剥蚀深度与束斑直径比小于1∶1时,由剥蚀深度/束斑直径引起的元素分馏效应可以忽略不计。基体效应研究表明, 50 μm与10 μm激光束斑下基体效应没有明显的差别。以NIST610为校准物质,Ca为内标元素, 10 μm束斑直径下GSD-1G、StHs6/80-G和NIST612中的36种微量元素分析结果与定值基本吻合,分析结果与定值基本匹配。综合考虑在10 μm的空间分辨率下,该技术可满足准确分析微量元素的要求。
关键词 激光剥蚀-电感耦合等离子体质谱; ArF准分子激光; 元素分馏; 基体效应; 空间分辨率
2015-12-22收稿;2016-03-16接受
本文系中国地质大调查项目(No.12120113021500)资助
E-mail:wangyaping@cags.ac.cn
1 引 言
近年来, 原位微区分析技术向准纳米尺度方向快速发展,如单矿物元素成像[1]、矿物生长环带分析[2]、微量元素扩散研究[3]、火山灰[4]、岩石漆[5]示踪以及鱼耳石[6]环境指示等方面的应用。随着分析区域趋向更小化,高空间分辨率分析将会越来越受到重视。激光剥蚀-电感耦合等离子体质谱法(LA-ICP-MS)作为一种重要的原位微区分析技术手段,具有空间分辨率高、检出限低等特点,广泛应用于分析地球化学领域[7~9]。目前元素分馏是制约该技术发展的主要瓶颈之一[10]。193 nm ArF准分子激光具有相对廉价、稳定性好等优点[11],在国内外LA-ICP-MS实验室占有较大比例。因此,研究此类型激光剥蚀系统的高空间分辨率元素分馏效应有重要意义及实用价值。
当前, LA-ICP-MS高空间分辨率(<15 μm)分析面临的主要问题有:灵敏度低、分析不确定度大、聚焦困难以及元素分馏严重等。范晨子等[12]的研究表明,当激光束斑直径为5 μm时,剥蚀坑呈椭圆形,这可能是由于激光聚焦问题导致。元素分馏主要来自3个方面[10]: (1)剥蚀过程的激光热效应和剥蚀深度/束斑直径比[13~15]; (2)传输过程中气溶胶传输速度不同和丢失[16,17]; (3)电感耦合等离子体离子化过程中ICP质量负载等因素[18]。高空间分辨率下由于激光束斑小及剥蚀量少等原因,剥蚀深度/束斑直径比与ICP质量负载元素分馏效应明显增加。Fryer等[19]最早定义了剥蚀过程中元素分馏指数(Fractionation Index,FI),以Ca为内标对元素信号进行标准化,剥蚀时间内后半段与前半段的比值。Kroslakova等[20]研究表明,随着ICP负载的增加(至16倍),元素信号比值(如Cu/Ca)降低超过25%。目前有关高空间分辨率下的元素分馏研究不多[21~23]。
在综合文献[4,22]研究结果基础上,本研究以国际标准物质为研究对象,研究了高空间分辨率下193 nm ArF准分子激光剥蚀系统的仪器检出限、ICP质量负载元素分馏、剥蚀深度/束斑直径元素分馏以及基体效应,分析了元素分馏机理,并在10 μm空间分辨率下准确分析了现有国际标准物质GSD-1G、StHs6/80-G和NIST612中的微量元素。
2 实验部分
2.1 实验仪器及调试
采用RESOlution M-50 型号193 nm ArF 准分子激光剥蚀系统(澳大利亚ASI公司)与Element 2 双聚焦扇形磁场等离子体质谱(美国Thermo Scientific公司)联用的LA-ICP-MS系统(哥廷根大学,德国)。仪器的主要工作参数选择见表1。以NIST 612作为校准物质,调节7Li, 139La和232Th信号最高,U/Th信号比约等于1,氧化物产率(ThO/Th)小于0.5%,二次离子产率(Ca2+/Ca+)小于0.5%。在7Li~238U质量范围内共扫描77种同位素,单次扫描总时间为1 s。
2.2 实验样品
实验样品均为国际标准物质,分别为美国标准技术研究院(NIST)合成玻璃标准物质 NIST 610和NIST 612,美国地质调查局USGS合成玻璃标准物质GSD-1G,德国马普化学研究所MPI-DING地质玻璃标准物质StHs6/80-G和ATHO-G。标准物质的定值及不确定度引自文献[24,25]以及GeoReM地质和环境分析标准物质数据库(http://georem.mpch-mainz.gwdg.de/)。样品表面抛光至1 μm,待测前采用乙醇超声清洗,氮气吹干。
2.3 剥蚀模式及数据处理
本实验采用点剥蚀和线扫描剥蚀两种剥蚀模式。点剥蚀模式,每个剥蚀点分析时间为220 s(剥蚀深度/束斑直径元素分馏研究)及75 s(基体效应研究),其中剥蚀前后空白采集时间均为20 s,剥蚀时间为180和35 s,剥蚀频率为5 Hz,总扫描次数220和75次。线扫描剥蚀模式,每条剥蚀线分析时间为75 s,其中剥蚀前后空白采集时间为20 s,剥蚀时间为35 s,剥蚀频率为10 Hz,线扫描速度为15 μm/s。
数据处理分别在Iolite 3.0和Microsoft Excel中进行,其中Iolite 3.0 进行仪器信号漂移校正和定量分析校准,Excel 进行元素分馏数据处理。
3 结果与讨论
3.1 高空间分辨率下的仪器检出限
仪器检出限与激光剥蚀参数(如样品剥蚀性质、束斑直径、能量密度及剥蚀频率等)密切相关。总体来说,单位时间内剥蚀出的物质越多,检出限越低。高分辩模式(< 15 μm)比普通模式(> 50 μm)检出限高。图1为采用NIST 612,激光能量密度3.0 J/cm2,剥蚀频率为5 Hz,在不同束斑直径(7, 10, 15, 33, 50和75 μm)的仪器检出限。从图1中可见,随着束斑直径减小,仪器检出限升高。当束斑直径大于50 μm时,大多数微量元素的仪器检出限小于0.01 μg/g;束斑直径降低为10 μm时,大多数微量元素的仪器检出限在0.1~1.0 μg/g; 而当束斑直径为7 μm时,一部分微量元素的仪器检出限在1.0~10.0 μg/g,已经不能很好地满足该技术在地球化学元素分析中的应用要求。为了降低高空间分辨率下LA-ICP-MS的检出限,可通过载气中添加少量H2或N2,以及采用屏蔽圈(Guard electrode)进行信号增敏[26~28]。
3.2 ICP质量负载元素分馏效应
ICP质量负载元素分馏是指由于进入炬管剥蚀物质量不同而引起的元素分馏。Li等[22]研究了193 nm ArF准分子激光剥蚀系统的ICP负载元素分馏效应,发现Cu/Ca、Zn/Ca和Pb/Ca 等元素信号比值随着负载的减小而增加,但其并未研究16 μm以下束斑的影响。本研究为了避免由于剥蚀深度/束斑直径引起的元素分馏,数据采集在线扫描的模式下进行,束斑直径分别为7, 10, 15, 33, 50和75 μm,分别代表不同剥蚀物质量。
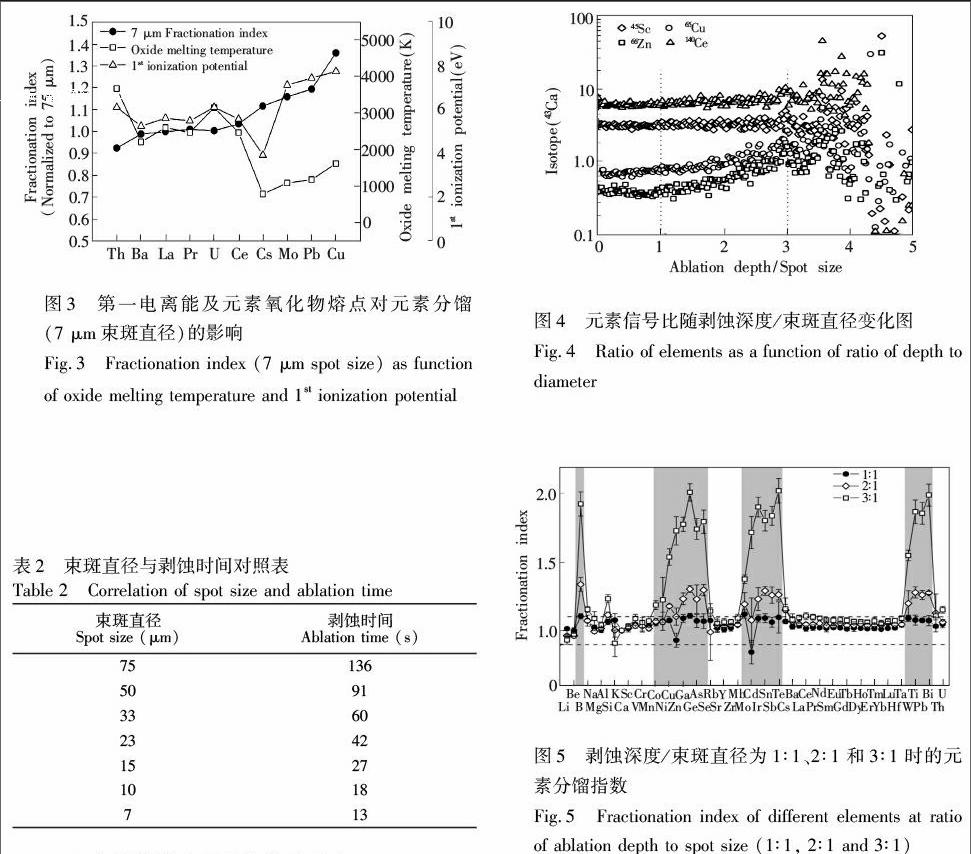
以43Ca为内标, 对所有元素信号强度进行标准化,考虑到小束斑下信号强度小,易造成较大不确定度,ICP质量负载元素分馏指数定义为不同束斑直径下元素/Ca与75 μm束斑直径下元素/Ca的比值。如图2所示,易挥发元素受到的影响大。随着剥蚀总量的减少,易挥发元素Cu, Ga, Mo, Cs及Pb的分馏指数,在剥蚀总量降低至19.36% (33 μm束斑直径)时保持不变,在4.00%~0.87%(15~7 μm)范围内逐渐增大,在低至0.87% (7 μm)时,元素分馏指数增大至1.15~1.35。 随着剥蚀总量的减少, 难熔元素如Ba, La及Ce等的分馏指数基本没有变化。这反映出剥蚀物在ICP炬管中未完全离子化[20],随着ICP质量负载的减少,易挥发元素相比于Ca离子化更完全。
进一步分析可知,ICP质量负载元素分馏主要受元素氧化物熔点和第一电离能控制[29]。从图3可见,当ICP质量负载降低至0.87%(7 μm)时,元素分馏指数和元素氧化物熔点呈负相关,表明氧化物熔点低的元素容易进行“原子化”。元素分馏指数与第一电离能有相同的趋势,说明ICP负载降低时,离子化效率提高,特别是对第一电离能较高的元素。
3.3 剥蚀深度/束斑直径元素分馏效应
剥蚀深度/束斑直径元素分馏一直是该分析技术的重要研究内容[30~32],并直接影响分析结果的准确性[33~35]。高空间分辨率下, 由于剥蚀束斑直径小,容易导致较严重的元素分馏。本研究遵循在保证足够信号强度并尽可能选择最小剥蚀束斑的原则,选用束斑直径23 μm,剥蚀频率5 Hz,剥蚀时间180 s等参数,研究了剥蚀深度/束斑直径的元素分馏行为。
采用光学显微镜(型号:Leica DMRX,徕卡显微系统(德国)有限公司)测量剥蚀坑直径和深度。经光学显微镜测量可知,激光能量密度3.0 J/cm2,玻璃标准物质的剥蚀速率为0.11 μm/脉冲。在不考虑随剥蚀深度增加引起的激光聚焦问题,连续剥蚀180 s后,剥蚀坑深度为99 μm。图4为元素/Ca随剥蚀深度/束斑直径的变化图, Cu/Ca和Zn/Ca随着剥蚀深度/束斑直径的增加而逐渐增大,而Sc/Ca和Ce/Ca基本保持不变。元素分馏指数定义为剥蚀时间内元素/Ca的后半段与前半段的比值[19]。图5为57种元素在剥蚀深度/束斑直径为1∶1, 2∶1和3∶1时的分馏指数, 在剥蚀深度/束斑直径为1∶1时,大多数元素分馏不明显; 但剥蚀深度/束斑直径增加至3∶1时,易挥发元素(B)、亲硫元素(Cu, Zn, Ga等)和亲铁元素(Co, Ni, W等)分馏趋势明显增加,亲石元素(Sc, Sr, REEs等)分馏趋势不明显,这与文献[19,21,31]报道一致。虽然元素分馏随剥蚀时间的增加趋向严重,但本实验结果表明,当剥蚀深度/束斑直径小于1∶1时, 绝大多数元素的分馏效应可以忽略不计。
在当激光能量密度为3.0 J/cm2, 剥蚀频率为5 Hz,束斑直径与剥蚀时间如表2所示时,剥蚀深度/束斑直径(<1∶1)引起的分馏效应可以忽略不计。
3.4 高空间分辨率下基体效应研究
基体效应是影响LA-ICP-MS分析结果准确性的主要因素之一,特别是当采用非基体匹配标准物质校准时[21,36,37]。袁继海等[38]提出了以分析元素与内标元素的信号比为纵坐标,浓度比为横坐标绘图,并以线性相关系数(R2)和相对灵敏度系数(Relative Sensitivity Factor, RSF)的相对标准偏差(RSD)描述基体效应。本研究采用NIST 610, GSD-1G, ATHO-G以及StHs6/80-G为研究对象,Ca为内标元素, 剥蚀束斑直径为10 μm,剥蚀时间为18 s,选择了9种元素,质量数从低到高,分别代表不同质量数的元素,研究了在高空间分辨率下的基体效应,如表3所示。
由表3可知,10 μm下的RSD(%)比50 μm大,这是因为小束斑下仪器信号不确定度较大等原因造成,与基体效应无关。在50和10 μm束斑直径下,绝大多数元素的RSF线性相关系数(R2)基本没有差别。表明高分辨模式(10 μm)与普通模式(50 μm)相比,基体效应没有明显改变。
3.5 10 μm束斑直径分析GSD-1G, StHs6/80-G 和NIST612
采用激光能量密度为3.0 J/cm2,剥蚀频率为5 Hz,剥蚀时间18 s等参数,以NIST610为校准物质,Ca为内标,在10 μm空间分辨率下,分析了GSD-1G、StHs6/80-G和NIST612中的36种微量元素,结果如表4所示。从表4可知,大多数元素测定浓度与定值浓度吻合,但GSD-1G中的Cr,Cu,Rb和Cs以及StHs6/80-G中的Rb的测定浓度较定值浓度偏高,NIST612中的Co偏低,这可能是由于分析不确定度较大造成的。综上分析,在本实验中选取的激光参数条件下,可在10 μm束斑直径下准确分析现有玻璃标准物质中的微量元素。
4 结 论
系统研究了高空间分辨率下193 nm ArF准分子激光剥蚀系统的仪器检出限、元素分馏以及基体效应。研究表明,仪器检出限随束斑直径的减小而升高,当束斑直径大于50 μm时,绝大多数微量元素的仪器检出限低于0.01 μg/g;束斑直径降低为7 μm时,部分微量元素的仪器检出限在1.0~10.0 μg/g范围,已经不能满足地球化学分析要求。ICP负载元素分馏效应与元素氧化物熔点和第一电离能密切相关,氧化物熔点低及第一电离能大的元素分馏现象明显。剥蚀深度/束斑直径元素分馏效应与剥蚀深度/束斑直径密切相关,当剥蚀深度与束斑直径小于1∶1时,元素分馏效应可以忽略不计。基体效应研究表明, 50与10 μm基体效应没有明显差别。在10 μm空间分辨率准确分析了GSD-1G、StHs6/80-G和NIST612中的36种微量元素,分析结果与定值基本匹配。随仪器增敏效应研究的发展,可以进一步提高LA-ICP-MS的空间分辨率。
References
1 Ubide T, McKenna C A, Chew D M, Kamber B S. Chem. Geol., 2015, 409: 157-168
2 Gerdes A, Zeh A. Chem. Geol., 2009, 261(3): 230-243
3 Selby D, Creaser R A. Geochim. Cosmochim. Acta, 2004, 68(19): 3897-3908
4 Tomlinson E L, Thordarson T, Müller W, Thirlwall M, Menzies M A. Chem. Geol., 2010, 279(3): 73-89
5 Macholdt D S, Jochum K P, Phlker C, Stoll B, Weis U, Weber B, Müller M, Kappl M, Buhre S, Kilcoyne A L D, Weigand M, Scholz D, Al-Amri A M, Andreae M O. Chem. Geol., 2015, 411: 57-68
6 Sanborn M, Telmer K. J. Anal. At. Spectrom., 2003, 18(10): 1231-1237
7 Liu Y S, Hu Z C, Li M, Gao S. Chinese Sci. Bull., 2013, 58(32): 3863-3878
8 Garbe-Schonberg D, Müller S. J. Anal. At. Spectrom., 2014, 29(6): 990-1000
9 KE Yu-Qiu, ZHANG Lu-Yuan, CAI Xin-Na, ZHENG Hong-Tao, JIN Lan-Lan, HU Sheng-Hong. Chem. J. Chinese Universities, 2012, 33(2): 257-262
柯于球, 张路远, 柴辛娜, 郑洪涛, 靳兰兰, 胡圣虹. 高等学校化学学报, 2012, 33(2): 257-262
10 Zhang S, He M, Yin Z, Zhu E, Hang W, Huang B. J. Anal. At. Spectrom., 2016, 31: 358-382
11 Müller W, Shelley M, Miller P, Broude S. J. Anal. At. Spectrom., 2009, 24(2): 209-214
12 FUN Chen-Zi, ZHAN Xiu-Chun, ZENG Pu-Sheng, HU Ming-Yue. Rock and Mineral Anal., 2015, 34(6): 609-616
范晨子, 詹秀春, 曾普胜, 胡明月. 岩矿测试, 2015, 34(6): 609-616
13 Guillong M, Horn I, Günther D. J. Anal. At. Spectrom., 2003, 18(10): 1224-1230
14 Machida R, Nakazawa T, Sakuraba Y, Fujiwara M, Furuta N. J. Anal. At. Spectrom., 2015, 30(12): 2412-2419
15 YUAN Ji-Hai, ZHAN Xiu-Chun, FAN Chen-Zi, ZHAO Ling-Hao, SUN Dong-Yang, JIA Ze-Rong, HU Ming-Yue, KUAI Li-Jun. Chinese J. Anal. Chem., 2012, 40(2): 201-207
袁继海, 詹秀春, 范晨子, 赵令浩, 孙冬阳, 贾泽荣, 胡明月, 蒯丽君. 分析化学, 2012, 40(2): 201-207
16 Koch J, Wlle M, Dietiker R, Günther D. Anal. Chem., 2008, 80(4): 915-921
17 Garcia C C, Lindner H, Niemax K. Spectrochim. Acta. Part B., 2007, 62(1): 13-19
18 Fietzke J, Frische M. J. Anal. At. Spectrom., 2016, 31(1): 234-244
19 Fryer B J, Jackson S E, Longerich H P. Canadian Mineralogist., 1995, 33: 303-303
20 Kroslakova I, Günther D. J. Anal. At. Spectrom., 2007, 22(1): 51-62
21 Hu Z C, Liu Y S, Chen L, Zhou L A, Li M, Zong K Q, Zhu L Y, Gao S. J. Anal. At. Spectrom., 2011, 26(2): 425-430
22 Li Z, Hu Z C, Liu Y, Gao S, Li M, Zong K, Chen H, Hu S. Chem. Geol., 2015, 400: 11-23
23 Fricker M B, Kutscher D, Aeschlimann B, Frommer J, Dietiker R, Bettmer J, Günther D. Inter. J. Mass. Spectrom., 2011, 307(1): 39-45
24 Jochum K P, Weis U, Stoll B, Kuzmin D, Yang Q C, Raczek I, Jacob D E, Stracke A, Birbaum K, Frick D A, Günther D, Enzweiler J. Geostand. Geoanal. Res., 2011, 35(4): 397-429
25 Jochum K P, Stoll B, Herwig K, Willbold M, Hofmann A W, Amini M, Aarburg S, Abouchami W, Hellebrand E, Mocek B, Raczek I, Stracke A, Alard O, Bouman C, Becker S, Dücking M, Brtz H, Klemd R, de Bruin D, Canil D, Cornell D, de Hoog C J, Dalpé C, Danyushevsky L, Eisenhauer A, Gao Y, Snow J E, Groschopf N, Günther D, Latkoczy C, Guillong M, Meixner A, Rosner M, Misawa K, Nash B P, Pfnder J, Premo W R, Sun W D, Tiepolo M, Vannucci R, Vennemann T, Wayne D, Woodhead J D. Geochemistry, Geophysics, Geosystems., 2006, 7(2): 1-44
26 Hu Z C, Gao S, Liu Y S, Hu S H, Chen H H, Yuan H L. J. Anal. At. Spectrom., 2008, 23(8): 1093-1101
27 Chen T, Hu Z C, Liu S, Liu Y, Gao S, Li M, Zong K, Chen H, Hu S. Spectrochim. Acta. Part B, 2015, 106: 36-44
28 Guillong M, Heinrich C A. J. Anal. At. Spectrom., 2007, 22(12): 1488-1494
29 Ho K S, Lee W W, Chan W T. J. Anal. At. Spectrom., 2015, 30(10): 2066-2073
30 Eggins S M, Kinsley L P J, Shelley J M G. Appl. Surf. Sci., 1998, 127-129: 278-286
31 Mank A J G, Mason P R D. J. Anal. At. Spectrom., 1999, 14(8): 1143-1153
32 Borisov O V, Mao X, Russo R E. Spectrochim. Acta. Part B, 2000, 55(11): 1693-1704
33 Longerich H, Günther D, Jackson S. Fresenius J. Anal. Chem., 1996, 355(5): 538-542
34 Kuhn H-R, Günther D. Anal. Chem., 2003, 75(4): 747-753
35 Luo T, Wang Y, Hu Z C, Günther D, Liu Y, Gao S, Li M, Hu S H. J. Anal. At. Spectrom., 2015, 30(4): 941-949
36 Jochum K P, Stoll B, Weis U, Jacob D E, Mertz-Kraus R, Andreae M O. Geostand. Geoanal. Res., 2014, 38(3): 265-292
37 YUAN Ji-Hai, ZHAN Xiu-Chun, SUN Dong-Yang, ZHAO Ling-Hao, FAN Chen-Zi, KUAI Li-Jun, HU Ming-Yue. ChineseJ. Anal. Chem., 2011, 39(10): 1582-1588
袁继海, 詹秀春, 孙冬阳, 赵令浩, 范晨子, 蒯丽君, 胡明月. 分析化学, 2011, 39(10): 1582-1588
38 YUAN Ji-Hai, ZHAN Xiu-Chun, HU Ming-Yue, ZHAO Ling-Hao, SUN Dong-Yang. Spectroscopy and Spectral Analsis., 2015, 35(2): 512-518
袁继海, 詹秀春, 胡明月, 赵令浩, 孙冬阳. 光谱学与光谱分析, 2015, 35(2): 512-518
Abstract Limits of detection (LODs), ICP mass load effect, downhole induced fractionation and matrix effect of 193 nm ArF excimer laser ablation system at high spatial resolution were systematically investigated. Trace elements in GSD-1G, StHs6/80-G and NIST612 were analyzed at 10 μm spot size. The results showed that LODs decreased with increasing ablation diameter. LODs of some trace elements were in a range of 1-10μg/g at 7 μm spot size. Mass load effect was negatively correlated with corresponding oxide melting temperature, while positively correlated with elemental 1st ionization potential. Downhole fractionation was negligible when the ratio of ablation depth versus spot size was smaller than 1∶1. Matrix effect based on elemental pair method showed that there was no significant change between spot sizes of 50 μm and 10 μm among investigated reference materials (NIST610, GSD-1G, ATHO-G and StHs6/80-G). Based on NIST610 as external standard and Ca as internal standard, the analytical results of 36 trace elements in GSD-1G, StHs6/80-G and NIST612 at 10μm spot size matched well with the reference value. In general, 10 μm spatial resolution could satisfy the requirements of trace element analysis。
Keywords Laser ablation-inductively coupled plasma-mass spectrometry; ArF excimer laser; Elemental fractionation; Matrix effect; Spatial resolution
- “博士县长”柴生芳去世引发热议
- 呼和浩特原政协主席自杀身亡
- 腾格里沙漠遭工业严重污染
- 李克强回信内蒙古村民谈“三农”
- 习近平:中西部强则中国强
- 四川将全面实施分级诊疗制度
- 贵州投入6800万元支持新型农业体
- 国家继续支持青海引大济湟工程建设
- 日喀则举行首届文化发展论坛
- 第九届丝绸之路市长论坛举行
- 丝绸之路经济带交通运输峰会举行
- 团中央确定今后三年409个援藏项目
- 习近平倡议打造中蒙俄经济走廊
- 习近平邀请印度总理到西安看玄奘译经处
- 罗永浩:有一种手机叫“锤子”
- 呼吁多建“既养老又看病”的养老院
- “不孝父母者暂缓入党”是一种正确导向
- 积极创新 务实探索
- 群众路线纾民困 清泉流淌在心间
- 激活“正能量” 汇聚“强动力”
- 两个古老文明的邂逅
- 郭氏宗亲祭祖掠影
- 米脂全方位堵塞“腐败源”
- 潼关县创建国家公共文化服务体系示范区
- 景俊海在安塞检查指导基层文化建设工作
- chortle
- chortled
- chortler
- chortlers
- chortles
- chortling
- chorus
- chorused
- choruses
- choruses'
- chorusing
- chorusses
- chose
- chosen
- chosenness
- chosenness'
- chosennesses
- chosenness's
- chosens
- choses'
- choses
- chow down
- christ
- christed
- christen
- 纱笼碧
- 纱纱
- 纱线
- 纱线扳牌楼——枉费心机
- 纱绢作布卖——不知好歹
- 纱绢当做粗布卖
- 纱绫
- 纱罗
- 纱罩
- 纱羊
- 纱衫
- 纱衾
- 纱貂底下没穷民
- 纱锭
- 纱雾
- 纲
- 纲一提则群目举,源一澄则众流清
- 纲上的总绳
- 纲举目张
- 纲举纪张
- 纲举网疏
- 纲宪
- 纲常
- 纲常大体
- 纲常扫地