肖坤 龚灿 郭强胜 许旭

摘 要 建立了定量核磁共振碳谱(13C-qNMR)技术测定食用油中特定位置不饱和脂肪酸含量的方法。使用反转门控去耦技术对食用油中的脂肪酸进行位置特异性分析,针对未完全分离的谱峰,比较了直接普通积分、使用不同的洛伦兹/高斯函数比值去卷积积分等数据处理方式对核磁共振碳谱定量结果的影响,选择以洛伦兹-高斯(3:2)的比例,对谱图进行去卷积拟合,提取sn-1,3和sn-2位亚油酸和油酸两种不饱和脂肪酸的定量峰,测定3种食用油中甘油三酯的饱和脂肪酸、sn-1,3位亚油酸、sn-2位亚油酸、sn-1,3位油酸、sn-2位油酸含量分别为:大豆油(16.2%、27.8%、24.0%、15.5%、7.9%(w/w,下同));玉米油(15.3%、30.7%、20.5%、20.1%、13.3%);花生油(18.3%、18.7%、12.5%、24.9%、25.5%)。大豆油中sn-1,3位亞麻酸、sn-2位亚麻酸分别为4.5%和4.0%,玉米油和花生油中未检出亚麻酸。上述结果与1H-qNMR测定的各脂肪酸总量一致。13C-qNMR可对食用油中脂肪酸进行位置特异性分析,无需复杂的样品前处理过程,可区分不饱和脂肪酸的不同位置分布,为在缺乏甘油三酯标样的情况下测量食用油中特定甘油三酯位置异构体成分含量提供了新方法。
关键词 定量核磁共振波谱法;核磁共振碳谱;食用油;脂肪酸;位置异构体
1 引 言
食用油是人体所需脂肪和能量的重要来源[1],也是体内必需脂肪酸的主要来源。目前,市场上常见的食用油多为植物油,如大豆油、玉米油、花生油和橄榄油等。甘油三酯(TAGs)是食用油的主要成分,因其甘油骨架上结合的脂肪酸不同而种类繁多[2],其中的亚麻酸、油酸、亚油酸以及棕榈酸等饱和脂肪酸的摄入量与多种慢性疾病有关[3~6]。食用油中脂肪酸的主要分析方法有气相色谱法(GC)[7~11]、气相色谱-质谱联用法(GC-MS)[12~14]等。这些方法需要复杂的样品前处理过程,即将甘油三酯转酯化为沸点较低的脂肪酸甲酯再分析[15]。Barison等[16]提出了一种简便的核磁共振氢谱(1H-NMR)方法,测定食用油中脂肪酸的相对含量,快速简便,且无需复杂的样品前处理过程,测定的脂肪酸结果与AOAC的气相色谱法一致。
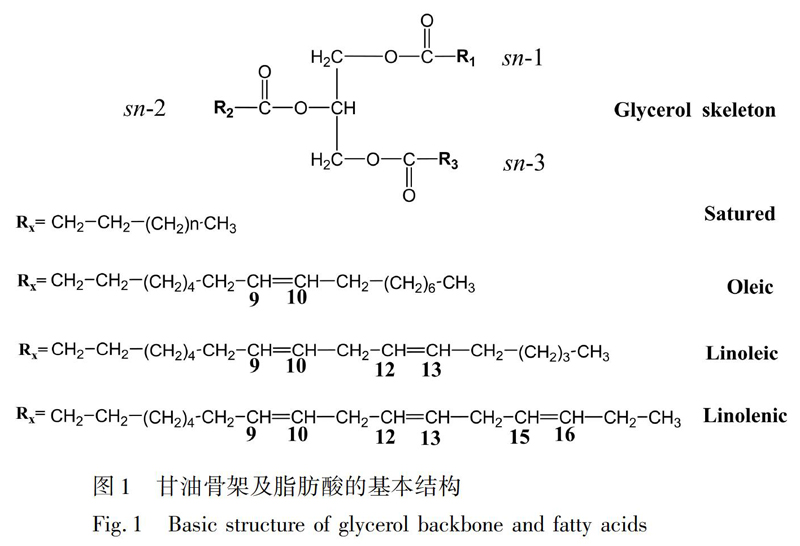
脂肪酸在甘油骨架中还存在位置特异性,可根据sn-1、sn-2、sn-3(sn: stereospecific numbering)编号区别各脂肪酸在甘油骨架的位置[7],其结构如图1所示。脂肪酸的位置对油脂的物理性质、生物化学性质和营养价值都有重要影响[17]。位于sn-2位的脂肪酸通过肠壁以甘油单酯的形式被吸收[18],可能对动脉粥样硬化、血脂和脂蛋白原形成不同的急性或慢性作用,并增加患心血管疾病的风险[19~21];而位于sn-1,3位的脂肪酸则作为游离的脂肪酸被人体吸收。研究表明[22,23],棕榈酸在甘油骨架的sn-2位具有促进婴幼儿对矿物质吸收的作用;而sn-1,3位的棕榈酸则很容易被肠道内脂肪酶水解成游离棕榈酸,与钙、镁等矿物质发生皂化反应,降低人体对脂肪的吸收利用效率。因此,对甘油三酯中的脂肪酸进行区域特异性分析非常重要。
脂肪酸在甘油骨架上的位置分析通常通过酶水解、格氏化学降解和色谱分析的组合确定[26]。常规的酶和化学方法存在长链多不饱和脂肪酸对某些脂肪酶催化分析的抗性[27]、通过格式化学降解可能引起的酰基迁移[28]、样品制备期间的损失或污染等缺点[29]。脂肪酸的核磁共振分析通常使用1H-NMR,但1H-NMR不能对脂肪酸进行位置特异性分析。13C-NMR的化学位移范围(约200 ppm)较大,可以识别几乎所有不饱和脂肪酸,而且可以通过特征化学位移区分不饱和脂肪酸的位置特异性,还可以避免甘油三酯的酶促和格氏水解中无法完全消除酰基迁移所引起的样品损失或污染等问题。因此,定量核磁共振碳谱(13C- qNMR)在分析食用油中不同位置脂肪酸时具有明显优势。
目前,13C-qNMR已被用于天然产物[30]和代谢物[31]的定量分析、材料科学[32]和脂质分析[33]等领域。甘油三酯骨架上的羰基碳是13C-qNMR的主要研究对象。然而,羰基碳自旋-晶格弛豫时间(T1)比其它类型的碳更长,而且脉冲角度、脉冲序列以及温度等实验参数对区域特异性分析影响较大,导致每次NMR实验时间较长[34]。这对于在较短的实验时间内实现最佳信噪比(S/N),进而进行准确的区域特异性分析提出了挑战。为缩短实验时间,Vlahov等[35]假设所有羰基碳受质子去耦的影响相同,在完全核Overhauser增强(NOE)下可获得信噪比较高的谱图。Suárez等[36]将宽带去耦脉冲序列应用于qNMR碳谱分析。然而,Gouk等[34]的研究表明,在sn-1,3和sn-2处的羰基碳的NOE可能不同,因此,宽带去耦脉冲序列并不能用于qNMR碳谱分析中。除了羰基碳,甘油三酯的sn-1,3和sn-2位的其它碳原子也具有不同的化学位移,如乙烯基碳。Meusel等[37]将GC、MALDI-TOF-MS和DEPT-45结合,通过分析乙烯基碳确定食用油中甘油三酯的含量。Merchak等[38]使用13C-INEPT序列(极化转移技术)和气相色谱(GC)分析橄榄油样品中脂肪酸和角鲨烯,同时用13C-INEPT光谱的去卷积峰面积作为预测因子构建多变量预测模型。相对复杂的HSQC-TOCSY NMR谱也已被用于分析甘油三酯混合物中棕榈酸和油酸的位置特异性分布[39]。通常,13C-NMR谱中的峰强度与碳核的数目不成正比,原因是每个碳原子的自旋-晶格弛豫时间不同,以及核的Overhauser增强(NOE)效应不同。然而,采用加顺磁弛豫试剂和加大脉冲间隔,特别是可以采用反转门控去耦消除NOE效应,同时消除全部质子的耦合,可以达到 13C核定量分析的目的[40]。脉冲傅里叶变换NMR的门控去偶(Gated decoupling)取13C的脉冲时间间隔tR> 5T1(T1为该化合物各碳原子中的最长纵向弛豫时间),这样可使磁化矢量恢复到平衡值,全去耦的碳谱NOE影响很小,谱线高度正比于碳原子数目,可用于定量分析。
本研究根据文献[16]的方法,以苯甲酸为内标,用1H-qNMR同时测定了大豆油中亚麻酸、亚油酸、油酸和饱和脂肪酸的绝对含量,进行了方法学验证。在此基础上,采用13C-NMR反转门控去耦技术对食用油中的脂肪酸进行位置特异性分析,应用去卷积法提取sn-1,3和sn-2位上油酸和亚油酸的峰面积,选择苯甲酸作为内标计算各位置不同脂肪酸的含量。本方法简便且无需复杂的样品前处理过程,可在没有甘油三酯标准样品的情况下,对食用油中甘油三酯的位置特异性脂肪酸进行绝对含量的分析测定。
2 实验部分
2.1 仪器与试剂
Bruker AVANCE III 500MHz核磁共振谱仪(德国Bruker公司);VGT-2120QT超声波清洗机(广东固特超声实业有限公司);DC-12氮吹仪(上海安谱科学仪器有限公司)。
苯甲酸(基准物质,99.95%~100.05%,国药集团化学试剂有限公司);氘代氯仿(99.8% D +0.03% TMS,Cambridge Isotope Lab公司);油酸(Oleic Acid,>99.0%)和亚油酸(Linoleic acid,>99.0%,日本Tokyo Chemical Industry公司);亚麻酸(Linolenic Acid,>99.0%,美国Acros Organics公司);三油酸甘油酯(OOO)、三亚油酸甘油酯(LLL)(上海阿拉丁试剂有限公司);1,2-二油酸-3-亚麻酸甘油三酯(OOLn)、1,3-二油酸-2-棕榈酸甘油三酯(OPO)(瑞典Larodan精细化学品有限公司)。 所有甘油三酯均冷冻保存。
大豆油(Soybean oil)、玉米油(Corn oil)和花生油(Peanut oil)购于本地超市。
2.2 样品的制备
称取约200 mg样品,溶于0.5 mL CDCl3,氮气吹扫2 min后,加入准确称取的苯甲酸内标,并超声2 min,去除样品中顺磁性溶解氧对实验的影响,然后转移至5 mm的核磁管中待用。所有油样均用60℃水浴加热10 min,确保其均匀性。
2.3 定量核磁共振碳谱
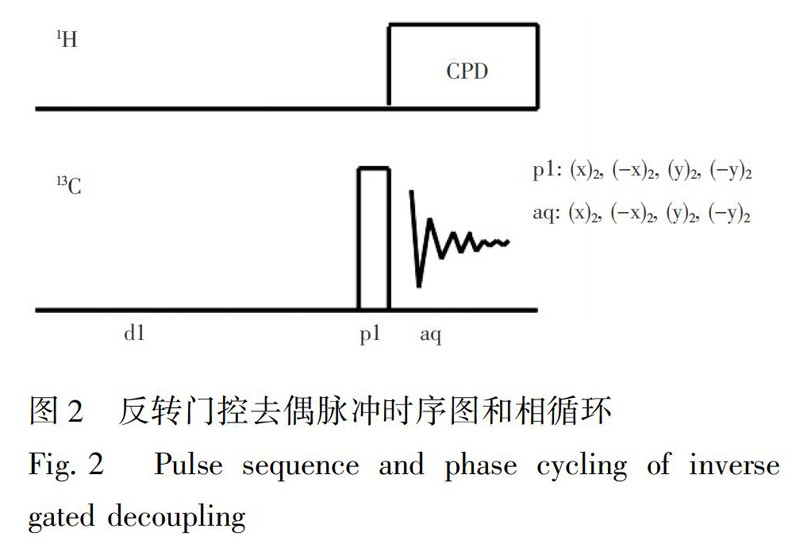
13C-qNMR采集条件:反转门控去耦脉冲序列zgig30,其脉冲时序图和相循环如图2所示。扫描次数NS=256,空扫次数DS=4,延迟时间D1 =35 s,谱宽SW=285 ppm,O1P=110 ppm,O2P=4.5 ppm,SI=256 K,测定NMR图谱。积分前先调平NMR基线,选择对应的峰的积分区间,每个峰积分3~5次,相对标准偏差(RSD)<1%时取平均值。在对谱图进行处理之前,先进行相位校正和基线调平。最后,根据中国药典的绝对定量公式计算待测组分的含量(Ws)[41]:
其中Wr为称取得苯甲酸(内标)的质量,As和Ar分别为供试品和苯甲酸内标峰(δ 129.43 ppm)的峰面积,Es和Er分别为供试品和苯甲酸内标的当量重量(分子量/該共振峰处的碳核数目)。
2.4 1H-qNMR谱中脂肪酸的分析
1H-qNMR采集条件:脉冲序列zg30。选择脉冲宽度P1=14.10 μs,延迟时间D1=6 s,扫描次数NS=32,测定NMR图谱。参考文献[16]中脂肪酸的计算方法,并加入内标苯甲酸,对大豆油中脂肪酸的含量进行测定。
3 结果与讨论
3.1 脂肪酸的1H-NMR分析
3.1.1 实验参数的影响 分别考察了不同延迟时间D1和扫描次数NS对1H-NMR谱积分面积的影响。当D1≥5 s,NS≥16时,信噪比(S/N)可以达到150以上,且Ar与As趋于稳定。故选择参数P1=14.10 μs,D1=6 s,NS=32。
3.1.2 方法学考察结果 亚麻酸、亚油酸、油酸3种脂肪酸线性相关系数(R)为0.9990~0.9997,日内精密度(RSD)为0.2%~0.7%,日间精密度(RSD)为0.8%~2.0%。根据中国药典的标准曲线剩余标准偏差和英国药典对qNMR要求(S/N ≥ 150:1),估计3种脂肪酸的检出限在1.4~5.5 g/L之间,定量限为4.8~17.2 g/L。在测定后的样品中加入3种脂肪酸(加标量均约为样品中含量的两倍),测得加样回收率为96.8%~102.9%。
3.1.3 样品中主要脂肪酸的测定 分别平行称取200 mg左右大豆油样品,用2.4节的方法的测定其中主要不饱和脂肪酸的含量(n=3)为:亚麻酸9.1%±0.6%、亚油酸51.5%±1.7%、油酸23.2%±1.5%、饱和脂肪酸16.2%±1.2%(w/w)。
3.2 脂肪酸的13C-qNMR分析
3.2.1 食用油中位置脂肪酸的13C-NMR峰归属 采用2.3节的方法,测定几种甘油三酯试剂纯品的NMR谱图,具体归属见图3。
对比图1中的甘油骨架和脂肪酸的基本结构,对比甘油三酯纯品的13C-NMR谱图,并参考文献[38],确定食用油中不同位置脂肪酸的13C-NMR化学位移如图4和图5所示。具体归属结果见表1。
3.2.2 13C核弛豫时间(T1)的测定
通过反转恢复脉冲序列测量了用于获得定量13C-NMR谱图的弛豫时间。选择脉冲序列: t1irpg,T1 delay设置为: 0.05、0.5、1、3、6、10、16、24和50 s。测得sn-1,3位亚油酸(L)的T1最大值为6.75 s。考虑脉冲延迟时间D1、弛豫时间T1与磁化矢量恢复关系。当D1为5倍T1时,磁化矢量恢复率大于99.9%。最终选择D1为35 s。
对于反转门控去偶技术,为了使去偶照射建立起来的NOE效应不影响下一次测定,必须增加两次NMR信号采样之间的延迟时间,但增加延迟时间会延长分析时间。通常在实验测定核弛豫时间(T1)的基础上,选择5倍T1或稍长的时间作为延迟时间。
3.2.3 13C-NMR谱定量峰的选择 根据图4中乙烯基碳的归属,在实际油样中选择δ=129.683 ppm(即图4中的G)和δ=129.657 ppm(即图4中的H)分别作为sn-1,3位油酸和sn-2位油酸的定量峰;δ=130.145 ppm(即图4中的A)和δ=130.139 ppm(即图4中的B)分别作为sn-2位亚油酸和sn-1,3位亚油酸的定量峰;除了大豆油中可以观察到亚麻酸sn-1,3和sn-2的位置特征峰,在其它3种食用油几乎观察不到亚麻酸的特征峰,因此,只对大豆油油中亚麻酸进行定量分析,δ=127.775 ppm(即图4中的Q)和δ=127.758 ppm(即图4中的R)分别作为sn-2位亚麻酸和sn-1,3位亚麻酸的定量峰。
根据图5中对羰基碳的归属,选择实际油样中δ=173.262 ppm(图5sn-1,3的O)和δ=172.887 ppm(即图5中sn-2的O)分别作为sn-1,3位油酸和sn-2位油酸的定量峰;δ=173.253 ppm(即图5中sn-1,3的L)和δ=172.947 ppm(即图5中sn-2的L)分别作为sn-1,3位亚油酸和sn-2位亚油酸的定量峰;对于饱和脂肪酸,选择δ=173.298 ppm(即图5中sn-1,3的P)作为其定量峰,在sn-2位置几乎检测不到饱和脂肪酸,因此,只对sn-1,3位的饱和脂肪酸进行定量分析。
3.2.4 13C-NMR谱的数据处理方式 当谱图中有峰重叠或受噪声影响过大时,可以使用去卷积方法算出各峰的峰面积。NMR峰的理论形状是由洛伦兹(Lorenz,L)方程给出的[42]。然而,由于场的不均匀性和加权函数可以产生部分高斯(Gauss,G)线形状。洛伦兹线性适用于底部较宽,顶部较尖的峰形,而高斯线性适用于底部较窄、顶部较缓的峰形。从图6可见,6B和6C是两个互补的形状,在某种程度上,大多数对称的峰可用洛伦兹和高斯线性以适当的比例近似代替。
本研究选择直接普通积分和去卷积积分两种积分方式对得到的碳谱谱图进行对比考察分析,其中,去卷积积分选择纯高斯、纯洛伦兹以及洛伦兹和高斯线性不同比例(1∶4,2∶3,1∶1,3∶2,3∶1)进行拟合。其中“普通积分”为选择两个波谷之间的部分进行直接中切法(Midcut method)积分处理。
由图6A(图3中的A和B两峰)可见,使用直接普通积分时,两个相邻峰的谱图重叠以及基线都会对峰面积造成影响,无法准确获取该化学位移处峰的面积信息,引起最终的结果的偏差。
去卷积积分,对需要去卷积的部分进行去卷积拟合,将得到的谱图用Bruker Topspin软件,Analysis菜单里的Line Ship Fitting→Lorenz/Gauss Deconvolution[dcon]功能处理,得到相应的峰面积。本研究选用7种去卷积拟合方式进行处理: 纯洛伦兹(b)、纯高斯(c)、洛伦兹∶高斯=1∶4(d)、2∶3(e)、1∶1(f)、3∶2(g)、3∶1(h),分析比较了去卷积结果,详见电子版文后支持信息表S1(乙烯基区域)和表S2(羰基区域)。当对谱图直接进行积分(中切法)时,得到的结果偏差较大。用纯洛伦兹或纯高斯进行去卷积拟合时,计算出的结果區别较大,两种峰去卷积拟合残差较大,与氢谱测定的脂肪酸总量仍有较大的差别。采用洛伦兹和高斯以一定比例进行去卷积拟合时发现,当洛伦兹∶高斯=3∶2时,拟合程度与原来的峰形最相似,且与氢谱测定的脂肪酸总量结果接近。 选择洛伦兹∶高斯=3∶2进行去卷积拟合提取。
对羰基区域的谱图进行同样处理,区域拟合的结果与氢谱计算得到的脂肪酸含量结果(饱和脂肪酸(16.2%±1.2%,亚油酸51.5%±1.7%,油酸23.2%±1.5%)接近。从表3可知,该区域拟合的结果同表2的结果接近。
考察了在洛伦兹∶高斯=60∶40(40% L)附近其它的比例。对于乙烯基区域,当洛伦兹∶高斯=62∶38(38% L)时,碳谱结果与氢谱最接近,对于羰基区域,洛伦兹∶高斯=58∶42(42% L)时,碳谱结果与氢谱最接近,考虑到整个图谱的处理方法尽量一致,且在40% L附近不同比例的差异不大。 选择以洛伦兹∶高斯=60∶40(40% L)的比例,同时用于去卷积计算乙烯基区域和羰基区域的峰面积。
大多数NMR峰可用洛伦兹和高斯以适当的比例近似。本研究选用洛伦兹∶高斯=3∶2进行去卷积拟合,对于不同的谱仪、仪器状态、样品,两者之间的比例可能也有差别,故在对谱图进行去卷积拟合时,建议通过考察实验数据选择合适的比例。
3.2.5 检出限(LOD) 根据中国药典四部[35]通则(2015版)规定,基于能显示基线噪声的分析方法,可以以信噪比为3或2时的相应浓度确定检出限(LOD)。本研究以普通光谱分析信噪比S/N>3确定检出限,选择本研究中采用的定量峰,根据其信噪比计算检出限,结果见表2。
对于qNMR,英国药典[103]要求根据S/N≥150确定定量限。对于碳谱,达到此信噪比的要求较难。中国药典四部[35]通则(2105版)中“9101 药品质量标准分析方法验证指导原则”以S/N=10时对应浓度为方法的定量限(LOQ)。上述研究结果可满足中国药典的信噪比要求。但较低的信噪比会使精密度差一些。需要进一步研究提高13C-qNMR信噪比和精密度的方法。本研究暂未对定量限做明确评价。
3.3 实际样品分析
称适量大豆油、玉米油和花生油样品和内标苯甲酸,按2.2节的方法配制成待测样品,用2.3节的方法测定。分析结果见表3,与3.1.3节中大豆油中脂肪酸的测定结果基本一致,说明NMR碳谱以内标法测定不同位置脂肪酸的结果可靠。
大豆油中亚油酸含量最多,且sn-1,3位亚油酸与sn-2位的亚油酸含量都高,其次是油酸,而亚麻酸含量较少,且这几种脂肪酸多位于sn-1,3位,饱和脂肪酸只在sn-1,3位。玉米油中亚油酸含量较高,且sn-1,3位较sn-2位含量多,花生油中油酸最多。由食用油的1H-qNMR分析结果可知,玉米油和花生油中含有的亚麻酸较少,在碳谱中难以观察到其特征峰,也难以测定其含量。而大豆油中的亚麻酸含量比较多,可采用13C-qNMR测定其含量。同时,在这几种食用油中,不饱和脂肪酸在sn-1,3位和sn-2位都有分布,并且含量相差不大,而饱和脂肪酸则都位于sn-1,3位,未检测到有sn-2位的饱和脂肪酸。
References
1 FANG Hong-Yun,HE Yu-Na,YU Dong-Mei,GUO Qi-Ya,WANG Xun,XU Xiao-Li,ZHAO Li-Yun. Food and Nutrition in China,2017,23(2): 56-58
房红芸,何宇纳,于冬梅,郭齐雅,王 寻,许晓丽,赵丽云. 中国食物与营养,2017,23(2): 56-58
2 ZHANG Yao-Li,PEI Xing-Li,GONG Can,HAN Yu-Liang,NI Tian-Qiang,WANG Fan,WANG Sheng-Jun,LU Hai-Peng,XU Xu. Chinese J. Anal. Chem.,2017,45(2): 183-190
张耀利,裴兴丽,龚 灿,韩玉良,倪天强,王 帆,王盛君,卢海鹏,许 旭. 分析化学,2017,45(2): 183-190
3 Helgadottir A,Gretarsdottir S,Thorleifsson G,Hjartarson E,Sigurdsson E,Magnusdottir A,Jonasdottir A,Kristjansson H,Sulem P,Oddsson A,Sveinbjornsson G,Steinthorsdottir V,Rafnar T,Masson G,Jonsdottir I,Olafsson I,Eyjolfsson G I,Sigurdardottir O,Daneshpour M S,Khalili D,Azizi F,Swinkels D W,Kiemeney L,Quyyumi A A,LeveyA I,Patel R S,Hayek S S,Gudmundsdottir I J,Thorgeirsson G,Thorsteinsdottir U,Gudbjartsson D F,Holm H,Stefansson K. Nat. Genetics,2016,48(6): 634-639
4 Risérus U,Willett W C,Hu F B. Prog. Lipid Res.,2009,48(1): 44-51
5 Houston D K,Ding J,Lee J S,Garcia M,Kanaya A M,Tylavsky F A,Newman A B,Visser N,Kritchevsky S B. Nutrit. Metab. Cardiovasc. Dis.,2011,21(6): 430-437
6 LI Jing,WANG Yong,YANG Yao-Dong,LEI Xin-Tao,XIAO Yong. Journal of Southern Agriculture,2016,47(12): 2124-2128
李 静,王 永,杨耀东,雷新涛,肖 勇. 南方农业学报,2016,47(12): 2124-2128
7 ZHU Tao-Hua,FAN Lu,QIAN Xiang-Ming,WANG Yan,JING Yin-Cheng. China Oils And Fats,2011,36(5): 59-63
朱桃花,范 璐,钱向明,王 艳,井银成. 中国油脂,2011,36 (5): 59-63
8 LI Bin,QIU Li-Qun,SONG Shao-Fang,GAO Ai-Ying,ZHANG Hao. Chinese Journal of Analysis Laboratory,2014,33(5): 528-532
李 斌,裘立群,宋少芳,高艾英,张 昊. 分析试验室,2014,33(5): 528-532
9 FAN Sheng-Xu,Li Bin,SUN Jun-Ming,HAN Fen-Xia,YAN Shu-Rong,WANG Lan,WANG Lian-Zheng. Chinese Journal of Oil Crop Sciences,2015,37(4): 548-553
范胜栩,李 斌,孙君明,韩粉霞,闫淑荣,王 岚,王连铮. 中国油料作物学报,2015,37(4): 548-553
10 FAN Jun-Ming,JIA Jun,CHEN Yan. Hubei Agricultural Sciences,2017,56(4): 727-730
凡军民,贾 君,陈 燕. 湖北农业科学,2017,56(4): 727-730
11 Haddad I,Mozzon M,Strabbioli R,Frega N G. Dairy Sci. Technol.,2012,92(1): 37-56
12 LOU Ting-Ting,ZHAO Ting,WANG Wei,LIN An-Qing,ZHAO Jing-Yuan. Food Research and Development,2014,35(21): 100-102
婁婷婷,赵 婷,王 伟,林安清,赵璟源. 食品研究与开发,2014,35(21): 100-102
13 LI Sha,WANG Wei-Xiang. Cereals & Oils,2015,28(9): 65-67
李 莎,王维香. 粮食与油脂,2015,28(9): 65-67
14 LIAO Li-Ping,XIAO Ai-Ping,LENG Juan,YANG Xi-Ai,LIU Liang-Liang,MEI Shi-Yong. China Oils and Fats,2017,42(4): 136-139
廖丽萍,肖爱平,冷 鹃,杨喜爱,刘亮亮,梅时勇. 中国油脂,2017,42(4): 136-139
15 Delmonte P,Kia A R F,Hu Q,Rader J I. J. AOAC Int.,2009,92(5): 1310-1326
16 Barison A,da Silva C W P,Campos F R,Simonelli F,Lenz C A,Ferreira A G. Mag. Reson. Chem.,2010,48(8): 642-650
17 Fauconnot L,Hau J,Aeschlimann J M,Fay L B,Dionisi F. Rapid Commun. Mass Spectrom.,2004,18(2): 218-224
18 Kayden H J,Senior J R,Mattson H F. J. Clin. Invest.,1967,46(11): 1695-1703
19 Myher J J,Marai L,Kuksis A,Kritchevsky D. Lipids,1977,12(10): 775-785
20 Manganaro F,Myher J J,Kuksis A,Kritchevsky D. Lipids,1981,16(7): 508-517
21 Berry S E E. Nutrit. Res. Rev.,2009,22(1): 3-17
22 BEI Pei,SUN Jian-Hua. International Journal of Pediatrics,2014,41(1): 58-60
贝 斐,孙建华. 国际儿科学杂志,2014,41(1): 58-60
23 SHANG Yun-Peng,SHENG Qing-Hai,WANG Zhen-Yu,ZHANG Zhi-Guo. Journal of the Chinese Cereals and Oils Association, 2010,25(10): 119-123
商允鹏,生庆海,王贞瑜,张志国. 中国粮油学报,2010,25(10): 119-123
24 Indelicato S,Bongiorno D,Pitonzo R,Stefano V D,Calabrese V,Indelicato S,Avellone G. J. Chromatogr. A,2017,1515: 1-16
25 Kim J,Jin G,Lee Y,Chun H S,Ahn S,Kim B H. J. Agric. Food Chem.,2015,63(40): 8955-8965
26 Gouk S U,Cheng S F,Ong A S H,Chuah C H. Eur. J. Lipid Sci. Technol.,2012,114(5): 510-519
27 Bottino N R,Vandenburg G A,Reiser R. Lipids,1967,2(6): 489-493
28 Becker C C,Rosenquist A,Holmer G. Lipids,1993,28(2): 147-149
29 Igarashi T,Aursand M,Hirata Y,Gribbestad I S,Wada S,Nonaka M. J. Am. Oil Chem. Soc.,2000,77(7): 737-748
30 Tenailleau E,Lancelin P,Robins R J,Akoka S. Anal. Chem.,2004,76(13): 3818-3825
31 Aursand M,Jorgensen L,Grasdalen H. Comp. Biochem. Physiol.,1995,112B(2): 315-321
32 Holtman K M,Chang H M,Jameel H,Kadla J F. J. Wood Chem. Technol.,2006,26(1): 21-34
33 Vlahov G,Giuliani A A,Re P D. Anal. Methods,2010,2(7): 916-923
34 Gouk S W,Cheng S F,Malon M,Onga A S H,Chuah C H. Anal. Methods,2013,5(8): 2064-2073
35 Vlahov G. Magn. Reson. Chem.,1998,36(5): 359-362
36 Suárez E R,Mugford P F,Rolle A J,Burton I W,Walter J A,Kralovec J A. J. Am. Oil Chem. Soc.,2010,87(12): 1425-1433
37 Meusel A,Popkova Y,Dannenberger D,Schiller J. Food Anal. Methods,2017,10(7): 2497-2506
38 Merchak N,Silvestre V,Loquet D,Rizk T,Akoka S,Bejjani J. Anal. Bioanal. Chem.,2017,409(1): 307-315
39 Simova S,Ivanova G,Passov S L. Chem. Phys. Lipids,2003,126(2): 167-176
40 YAN Bao-Zhen. Graphical Nuclear Magnetic Resonance Technology and Examples. Beijing: Science Press,2010: 240
严宝珍. 图解核磁共振技术与实例. 北京: 科学出版社,2010: 240
41 National Pharmacopoeia Committee. The Pharmacopoeia of the People's Republic of China ( Part IV,2015 Ed),Beijing: China Medical Science Press,2015: 52-54,376
國家药典委员会编. 中华人民共和国药典 (四部,2015年版). 北京: 中国医药科技出版社,2015: 52-54,376
42 GAO Han-Bin,ZHANG Zhen-FANG. Principles and Experimental Methods of Nuclear Magnetic Resonance. Wuhan: Wuhan University Press,2008: 25
高汉宾,张振芳. 核磁共振原理与实验方法. 武汉: 武汉大学出版社,2008: 25
- 依托本土文化开发幼儿园自主课程(美术领域)的研究
- 刍议民间乡土资源对幼儿发展的重要作用
- 浅论中小学后勤环境建设助推学生核心素养的培养
- 协同创新视域下大学生创新创业教育模式改革研究
- 农村小规模学校有效教学策略研究
- 建立科学合理的学校艺术教育评价制度研究
- 微视频在机器人教学中的学习行为研究
- 试论基于工匠精神引领下的中等职业学校德育教育
- 高职院校城市轨道交通工程技术专业人才培养方案研究
- 探究无止境 实践悟真知
- 校企合作背景下高职药学专业人才培养模式改革探索
- “小讲课”在基础护理教学中的尝试
- 无机非金属材料专业应用型人才的培养策略
- 基于“导师制”背景下的医学生医德教育探索
- 基于广东高职特色的人文素质课程开发的思考与实践
- 基于项目驱动法的应用型本科《工程概预算》课程教学方法初探
- 初探幼儿游戏活动评价的有效性
- 政府“放管服”视域下的高校学生管理模式探析
- 互联网+家校共育之初探
- STEAM教育应用于通用技术课程的可行性分析
- 乡土资源在农村幼儿园教学活动中的应用
- 社区教育资源开发的长效机制探析
- 中职学生核心素养培育初探
- 综合实践课程生活化情境教学模式探究
- 浅议幼儿园教育中游戏教学观察能力提升路径
- airline's
- airlock
- airlocks
- air-mail
- airmail
- airmailed
- air-mailed
- airmailing
- air-mailing
- air-mails
- airmails
- airman
- airmen
- airmiles
- air miles™
- air-out
- airplane
- airplanes
- airplane's
- air pocket
- air pockets
- airport
- airports
- air raid
- air raiders
- 扬气
- 扬水
- 扬汤止沸
- 扬汤止沸,莫如去薪
- 扬汤止沸,莫若去薪
- 扬汰
- 扬沙
- 扬沙走石
- 扬沙起石
- 扬波
- 扬流
- 扬浇
- 扬清
- 扬清抑浊
- 扬清激浊
- 扬灵
- 扬烈
- 扬煇
- 扬琴
- 扬白
- 扬白眼
- 扬眉
- 扬眉伸气
- 扬眉吐气
- 扬眉奋髯