杂质延迟液相色谱四极杆/离子阱复合质谱测定水产加工食品中23种全氟烷基化合物
郭萌萌等

摘 要 采用杂质延迟法去除液相系统中的背景干扰,利用液相色谱四极杆/线性离子阱复合质谱(LCMS/MSQTRAP)的同时定性定量功能,建立了水产加工食品中23种全氟烷基化合物(Perfluorinated alkyl substances, PFASs)的定性确证和定量测定方法。样品经酸化乙腈提取,C18填料和石墨化碳黑(GCB)分散固相萃取净化,C18色谱柱分离,甲醇和5 mmol/L乙酸铵溶液梯度洗脱;在液相系统混合器和进样器之间串联一根延迟色谱柱,去除液相系统的背景干扰;质谱采集使用MS/MSQTRAP独有的多反应监测(MRM)信息依赖性采集(IDA)增强子离子扫描(EPI)模式;同位素内标定量,在线EPI谱库定性确证。23种目标物在各自相应浓度范围内线性良好,相关系数不低于0.995,定量限为0.02~0.1 μg/kg。基质加标回收率在67.5%~116.4%之间,相对标准偏差(RSD)为5.2%~14.7%。本方法有效控制了液相系统的背景干扰,一次进样即可完成23种PFASs的确证和测定,适用于水产加工食品中PFASs的监控分析
关键词 全氟烷基化合物; 水产加工食品; 杂质延迟法; 液相色谱三重四极杆/线性离子阱复合质谱
全氟烷基化合物(Perfluoroalkylated substances,PFASs)是一类新型持久性有机污染物(Persistent Organic Pollutants,POPs),具有疏油、疏水特性,对物理、化学和生物降解均有较好的抵抗作用。研究表明,PFASs在多种环境介质和生物机体中广泛分布并持久性存在,且具有生殖毒性[1]、神经毒性[2]和免疫毒性[3]。2009年,全氟辛烷磺酸(盐)及全氟辛基磺酰氟被列入《斯德哥尔摩公约》优控名单[4];2010年欧盟发布2010/161/EU号[5]议案,提议开展食品中PFASs的监控;2011年,欧盟发布食品中PFASs污染的调查报告,发现水产动物源性食品中PFASs残留水平明显高于陆源食品,故而提出水产动物源性食品常态化监控PFASs的建议[6]。目前,我国仅对鲜活水产品中PFASs污染状况进行了初步调查,水产加工食品中PFASs污染仍不明确。比较而言,水产加工食品因增加了加工环节,加工过程、包装材料等均可能增加PFASs污染风险,另外鱼罐头、即食海产品等作为日常膳食的一类食品,是我国广大内陆地区消费者摄取水产蛋白的主要来源之一,因此建立水产加工食品中PFASs的检测方法十分必要。
目前,针对水产品中PFASs的检测方法多采用液相色谱串联四极杆质谱法进行分析[7~9],固相萃取法进行样品净化[9~11]。液相色谱串联四极杆质谱测定时,流动相溶剂、在线脱气机及色谱流路中均可能含有氟聚合物而引入背景干扰,部分组分如全氟己酸、全氟庚酸和全氟辛酸等化合物的仪器背景干扰值接近或高于定量限,难以实现PFASs的准确定量和有效监测;同时,水产加工食品的基质较鲜活水产品更为复杂,当样品中目标物残留较低时,采用四极杆质谱定性时部分化合物的两对MRM离子比率偏差较大,造成定性困难;另外,固相萃取净化方法的操作较繁琐,且对长碳链(C≥11)羧酸类PFASs的回收率不理想。针对上述问题,本研究结合杂质延迟技术,在液相系统混合器和进样器之间串联一根延迟色谱柱,去除了液相系统的背景值,并采用Qtrap质谱的多反应监测(MRM)信息依赖采集(IDA)增强子离子扫描(EPI)模式,实现了复杂基质中23种PFASs的同时定量定性分析,同时采用C18填料和石墨化碳黑(GCB)进行分散固相萃取净化,简化了样品前处理步骤,在保证全部目标物获得良好回收率的前提下,有效降低了复杂基质的干扰。这些改进显著提高了方法的准确性和可靠性,为准确监测水产加工食品中的PFASs污染水平提供了有效的技术保障。
2 实验部分
2.1 仪器、试剂与材料
Prominence UFLC液相色谱(Shimadzu公司);5500QTRAP四极杆线性离子阱复合质谱(AB SCIEX公司);T18 basic均质机(IKA公司);XW80A旋涡混合器(上海医大仪器厂);Himac CR 22GⅡ高速离心机(Hitachi公司);NEVAP 112氮吹仪(Organomation公司);MilliQ超纯水仪(Millipore公司)。
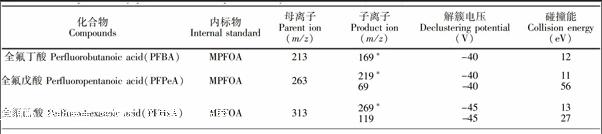
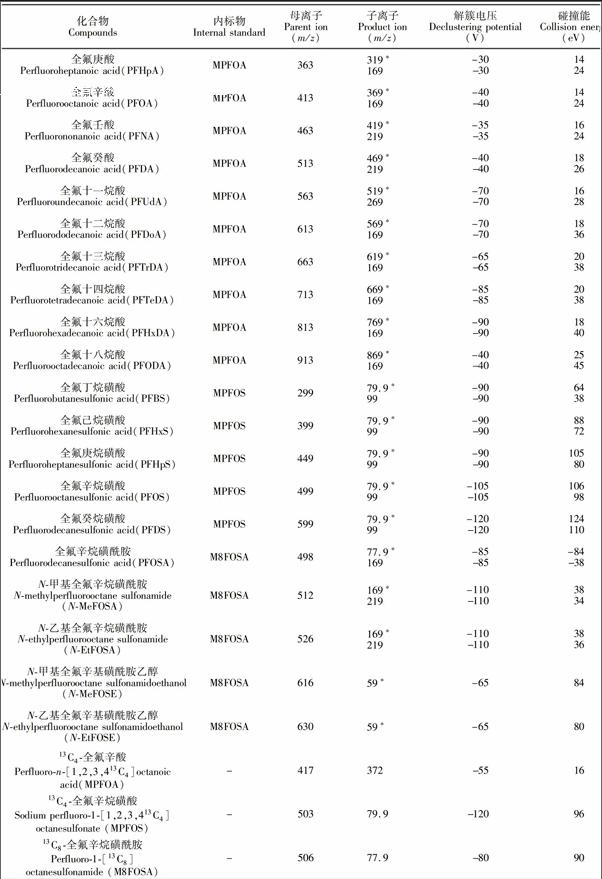
全氟丁酸、全氟戊酸、全氟己酸、全氟庚酸、全氟辛酸、全氟壬酸、全氟癸酸、全氟十一烷酸、全氟十二烷酸、全氟十三烷酸、全氟十四烷酸、全氟十六烷酸、全氟十八烷酸、全氟丁烷磺酸、全氟己烷磺酸、全氟庚烷磺酸、全氟辛烷磺酸、全氟癸烷磺酸、全氟辛烷磺酰胺、N甲基全氟辛烷磺酰胺、N乙基全氟辛烷磺酰胺、N甲基全氟辛基磺酰胺乙醇、N乙基全氟辛基磺酰胺乙醇、13C4全氟辛酸、13C4全氟辛烷磺酸、13C8全氟辛烷磺酰胺(Wellington Labortories公司);甲醇、乙腈(HPLC级,Merk公司);甲酸、乙酸铵(HPLC级,Fluka公司);超纯水(18.2 MΩ·cm); C18萃取剂(50 μm,艾杰尔公司);石墨化碳黑(CNWBOND CarbonGCB, 120~400目,CNW公司),其它未作特殊说明的试剂均为分析纯。
2.4 质量控制
为防止样品前处理步骤中引入高背景值,实验过程避免使用聚四氟乙烯材质的器皿,且器皿在使用前用超纯水和甲醇充分清洗。定量分析采用标准曲线校正,同位素内标法定量。每批样品进行测试时,同时做空白试验、基质加标试验和线性试验,空白试验确保试剂空白中所有分析物的含量低于定量限;基质加标试验加入PFASs标准物质(2 ng)和内标物(1 ng)验证方法的准确度,回收率控制在60%~120%范围内;采用至少5个浓度点进行标准曲线的绘制,且线性相关系数达0.99以上。
3 结果与讨论
3.1 杂质延迟法色谱条件的优化
依据色谱柱对液相系统中干扰物能捕集并释放的原理[11],流动相溶剂、在线脱气机和色谱流路中的PFASs在分析色谱柱中浓缩,可能与作为分析样品进样的目标物在同一时间检出。为了将来自液相系统的PFASs背景干扰组分与样品中的目标组分PFASs分离,在混合器和进样器之间安装延迟色谱柱(如图1),使干扰组分较目标组分延迟洗脱。
选用对PFASs有一定保留的色谱柱作为延迟柱[13],在液相系统混合器和进样器之间串联后,调整色谱的梯度洗脱条件, 使干扰组分在目标组分之后洗脱时基线分离(图2),达到了去除液相系统中背景干扰的目的。
3.2 质谱条件的优化
欧盟规定[14]用LCMS/MS方法检测有机污染物时,其定性确证方法要求每种化合物选用两对MRM离子,需要4个确证点(Identification Point,IP),即一个母离子,两个子离子(母离子IP为1.0,子离子IP为1.5)。但在本课题组前期建立的方法[7]中, PFBA, PFPeA, PFHxA, PFOSA, NMeFOSE和NEtFOSE仅能检测到一对稳定的MRM离子,且水产加工食品基质复杂,存在一定程度的基质抑制效应,当样品中目标物残留很低时,可能其它化合物的两对MRM离子也不能同时检测到,为定性分析带来极大困难。本方法首先采用目标物的单标溶液通过流动注射进质谱,以多反应监测负离子模式重点优化各化合物的子离子和碰撞能,增加了PFPeA, PFHxA和PFOSA的MRM扫描离子(见表1);然后利用MRMIDAEPI模式, 采用23种化合物的混合标准溶液通过自动进样器采集数据,该模式按照常规定量方法设定目标物的MRM离子对,在实际扫描过程中,尚某MRM通道采集的信号强度超过预设值后,就会触发EPI增强子离子扫描模式,进而获得对应MRM通道的母离子的增强二级离子全扫描质谱图,以用于定性分析; 同时建立目标物的在线EPI质谱库, 辅助定性。而此时,MRM通道采集的信号依然可以作为定量分析的数据。与MRM模式相比,EPI谱图由离子阱采集,其灵敏度高于四极杆,增强了二级碎片子离子扫描,且为全扫描质谱图,确证信息更为丰富,因而可以有效辅助痕量水平(例如≤0.1 μg/kg)或检出限浓度样品的定性确证,很好地解决了传统四极杆MRM模式对低浓度样品的定性难题。图3显示的是MRMIDAEPI模式与传统MRM模式下,0.1 μg/kg PFOA阳性沙丁鱼罐头样品的离子碎片信息。从图3可见,EPI扫描模式下,碎片离子响应增强近10倍,且该模式除采集母离子m/z 413和子离子m/z 369, 169外,还采集了m/z 219, 395等子离子的信息,确证点IP达7.0,远高于欧盟对有机污染物的确证要求,极大地提高了分析结果的可靠性。
3.3 样品前处理方法的优化
水产品加工食品的基质复杂,除脂肪、蛋白等内源性杂质外,还存在色素、糖等添加剂,本研究将鲜活水产品中PFASs的前处理方法[7]进一步优化,匀浆试样的取样量由5 g减为2 g,同时重点考察了净化填料用量的不同对目标物回收率和净化效果的影响。实验选取墨鱼丸样品为测试基质,按 2.2.1步骤进行提取,再添加2 ng PFASs标准物质,采用C18萃取剂吸附脂肪等非极性共萃物,通过对比50~250 mg C18对回收率和净化效果的影响,发现C18用量为150 mg时,平均回收率均优于其它用量(图4),且净化后的样品溶液澄清,去脂效果较好。在此基础上,添加GCB去除色素及具有共轭结构的谷氨酸、精氨酸等水产加工食品的风味成分,实验考察了150 mg C18与30~60 mg GCB的配比情况,GCB为50 mg时,可有效去除色素等杂质,且平均回收率高于90%(图5)。因此,本方法最终采用150 mg C18+50 mg GCB对样品提取液净化,在保证回收率的前提下,可有效降低水产加工食品中非极性和极性风味成分及脂肪酸等对测定的干扰。
3.4 灵敏度、准确度和精密度
取适量PFASs混合标准溶液和内标溶液,配制一系列浓度梯度的标准溶液,在2.3节条件下依次测定,以各组分和内标物的面积比值为纵坐标,各组分的质量浓度为横坐标进行线性回归分析。采用杂质延迟法,仪器的背景干扰得到了有效控制,5次平行测定中干扰组分的平均浓度不高于0.1 μg/L。在空白样品中添加低浓度的标准溶液,按2.2节步骤进行样品前处理后进样测定,以信噪比(S/N)≥10确定各组分定量限(LOQ),结果见表2。结果表明,23种化合物的线性良好,定量限为0.02~0.10 μg/kg,满足甚至高于目前食品基质中PFASs测定方法的指标[15,16]。
选取鱼丸、海米和沙丁鱼罐头为测试基质,分别添加相当于0.1, 1.0和10.0 μg/kg浓度的PFASs混合标准溶液进行测定,每个浓度做6个平行样品。同时做空白试验,扣除本底值后计算加标回收率和相对标准偏差。结果显示,PFASs在鱼丸中的加标回收率在72.2%~108.1%之间,相对标准偏差为5.2%~12.8%;PFASs在海米中的加标回收率在75.1%~116.4%之间,相对标准偏差为7.2%~13.5%;PFASs在沙丁鱼罐头中的加标回收率在67.5%~104.6%之间,相对标准偏差为5.9%~14.7%,方法的准确度与精密度良好。
3.5 实际样品分析
应用本方法分析了20份市售水产加工食品(鱼丸、虾丸、海米和沙丁鱼罐头各5份),发现部分样品存在PFASs污染(见表3),总量为0.82~7.437 μg/kg,并对阳性样品进行EPI谱库检索,匹配度均大于90%,且阳性样品中PFOA,PFOS和PFOSA的检出率较高,在样品中富集普遍。因此,建议开展水产加工食品中PFASs的残留监测,并以PFOA、PFOS和PFOSA为重点监控目标。
4 结 论
利用杂质延迟技术,通过在液相系统混合器和进样器之间安装延迟色谱柱,实现了色谱流路中PFASs假阳性干扰的去除。不过,由于延迟了目标物的保留时间,造成分析的平衡时间略有延长,但液相色谱系统的背景干扰得到有效控制,从而提高了方法的准确性;采用Qtrap质谱特有的MRMIDAEPI扫描模式实现一次进样同时定性与定量分析,有效判别假阳性样品,并解决痕量水平或检出限浓度样品的定性难题,提高了分析结果的可靠性;分散固相萃取的净化方式,简化了样品的前处理操作,降低了水产加工食品中风味成分及脂肪酸对测定的干扰。本方法的建立为进一步开展全国水产动物源性食品中全氟烷基化合物的残留水平调查提供了技术手段。
References
1 Wei Y H, Dai J Y, Liu M,Wang J S, Xu M Q, Zha J M, Wang Z J. Environ. Toxicol. Chem., 2007, 26(11): 2440-2447
2 Johansson N,Fredriksson A, Eriksson P. Neurotoxicology, 2008, 29(1): 160-169
3 Zheng L, Dong G H, Jin Y H, He Q C. Arch. Toxicol., 2009, 83: 679-689
4 WANG YaWei, CAI YaQi, JIANG GuiBin. Sci. Sin. Chim., 2012, 40(2): 99-123
王亚韡, 蔡亚岐, 江桂斌. 中国科学: 化学, 2012, 40(2): 99-123
5 Commission regulation (EC) No 2010/161/EU. Official Journal of the European Union, 2010
6 European Food Safety Authority.The EFSA Journal, 2011, 9(2): 2016
7 GUO MengMeng, WU HaiYan, LI ZhaoXin, TAN ZhiJun, ZHAI YuXiu. Chinese J. Anal. Chem., 2013, 41(9): 1322-1327
郭萌萌, 吴海燕, 李兆新, 谭志军, 翟毓秀. 分析化学, 2013, 41(9): 1322-1327
8 Wu Y N, Wang Y X, Li J G, Zhao Y F,Guo F F, Liu J Y, Cai Z W. Environ. Int., 2012, 42: 67-71
9 LAN Fang, FENG Sha, SHEN JinCan, WU XiaoPing, YUE ZhenFeng. Chinese J. Anal. Chem.,2013, 41(12): 1893-1898
蓝 芳, 冯 沙, 沈金灿, 吴晓萍, 岳振峰. 分析化学, 2013, 41(12): 1893-1898
10 Delinsky A D, Strynar M J, Nakayama S F, Varns J L, Ye X B, McCann P J, Lindstrom A B. Environ. Res., 2009, 109: 975-984
11 HE XiaoMin, CHEN Hao. Food Sci., 2014, 35(8): 193-197
贺小敏, 陈 浩. 食品科学, 2014, 35(8): 193-197
12 ZHANG XiuLan, GUO Jing, LI LingLing, DONG Liang, SHI ShuangXin, ZHANG LiFei, ZHOU Li, YANG WenLong, HUANG XingRu. Chinese J. Anal. Chem., 2014, 42(3): 452-456
张秀蓝, 郭 婧, 李玲玲, 董 亮, 史双昕, 张利飞, 周 丽, 杨文龙, 黄兴茹. 分析化学, 2014, 42(3): 452-456
13 Lacina O, Hradkova P, Pulkrabova J, Hajslova J. J. Chromatogr. A, 2011, 1218(28): 4312-4321
14 Commission Decision 2002/657/EC. Off. J. Eur. Commun., 2002, L221: 8
15 Hradkova P, Poustka J, louskova V, Pulkrabova J, Tomaniova M, Hajslova J. Czech. J. Food Sci., 2010, 28(4): 333-342
16 LI Jing, ZHANG Hong, CHAI ZhiFang, SHEN JinCan, YANG Bo. J. Instru. Anal., 2014, 33(10): 1109-1115
李 静, 张 鸿, 柴之芳, 沈金灿, 杨 波. 分析测试学报, 2014, 33(10): 1109-1115
act A comprehensive method for the simultaneous identification and detection of 23 perfluorinated alkyl substances (PFASs) in processed aquatic products by impurity delay was developed using liquid chromatography coupled with quadrupole/linear ion trap tandem mass spectrometry (LCQ/TrapMS). The sample was extracted with acidified acetonitrile, cleanedup by dispersive solid phase extraction using C18 and graphitized carbon blacks (GCB). The separation of 23 PFASs was performed on a Kinetex XBC18 (100 mm×2.1 mm,2.6 μm) column using gradient elution of 5 mmol/L ammonium acetate and methanol as mobile phase. And a short C18 HPLC column was inserted between the mixer and the autosample, which delayed compounds coming from the LC system. A scheduled multiple reaction monitoring (MRM) in negative mode as survey scan and an enhanced product ion (EPI) scan as dependent scan in an informationdependent acquisition (IDA) experiment were adopted in mass spectrometry acquisition. Online labbuilt MS/MS library and the isotope internal standards were employed for the identification and quantification. The calibration curves for the detection of 23 PFASs were linear well with correlation coefficient over 0.995. The limits of quantification for all analytes were ranged from 0.02 μg/kg to 0.1 μg/kg. The average spiked recoveries for 23 PFASs were between 67.5% and 116.4%, with relative standard deviations (RSDs) from 5.2% to 14.7%. The background coming from the part of LC was controlled well by the impurity delay. The proposed method can be used to identify and detect the 23 PFASs in a single run, and also suitable for the analysis of processed aquatic product samples.
Keywords Perfluorinated alkyl substances; Processed aquatic products; Impurity delay; Liquid chromatographyquadrupole/linear ion trap tandem mass spectrometry
(Received 5 February 2015; accepted 12 June 2015)
This work was supported by the Special Foundation for Basic Research Program of the Ministry of Science and Technology, China (No.20144FY30100)
摘 要 采用杂质延迟法去除液相系统中的背景干扰,利用液相色谱四极杆/线性离子阱复合质谱(LCMS/MSQTRAP)的同时定性定量功能,建立了水产加工食品中23种全氟烷基化合物(Perfluorinated alkyl substances, PFASs)的定性确证和定量测定方法。样品经酸化乙腈提取,C18填料和石墨化碳黑(GCB)分散固相萃取净化,C18色谱柱分离,甲醇和5 mmol/L乙酸铵溶液梯度洗脱;在液相系统混合器和进样器之间串联一根延迟色谱柱,去除液相系统的背景干扰;质谱采集使用MS/MSQTRAP独有的多反应监测(MRM)信息依赖性采集(IDA)增强子离子扫描(EPI)模式;同位素内标定量,在线EPI谱库定性确证。23种目标物在各自相应浓度范围内线性良好,相关系数不低于0.995,定量限为0.02~0.1 μg/kg。基质加标回收率在67.5%~116.4%之间,相对标准偏差(RSD)为5.2%~14.7%。本方法有效控制了液相系统的背景干扰,一次进样即可完成23种PFASs的确证和测定,适用于水产加工食品中PFASs的监控分析
关键词 全氟烷基化合物; 水产加工食品; 杂质延迟法; 液相色谱三重四极杆/线性离子阱复合质谱
全氟烷基化合物(Perfluoroalkylated substances,PFASs)是一类新型持久性有机污染物(Persistent Organic Pollutants,POPs),具有疏油、疏水特性,对物理、化学和生物降解均有较好的抵抗作用。研究表明,PFASs在多种环境介质和生物机体中广泛分布并持久性存在,且具有生殖毒性[1]、神经毒性[2]和免疫毒性[3]。2009年,全氟辛烷磺酸(盐)及全氟辛基磺酰氟被列入《斯德哥尔摩公约》优控名单[4];2010年欧盟发布2010/161/EU号[5]议案,提议开展食品中PFASs的监控;2011年,欧盟发布食品中PFASs污染的调查报告,发现水产动物源性食品中PFASs残留水平明显高于陆源食品,故而提出水产动物源性食品常态化监控PFASs的建议[6]。目前,我国仅对鲜活水产品中PFASs污染状况进行了初步调查,水产加工食品中PFASs污染仍不明确。比较而言,水产加工食品因增加了加工环节,加工过程、包装材料等均可能增加PFASs污染风险,另外鱼罐头、即食海产品等作为日常膳食的一类食品,是我国广大内陆地区消费者摄取水产蛋白的主要来源之一,因此建立水产加工食品中PFASs的检测方法十分必要。
目前,针对水产品中PFASs的检测方法多采用液相色谱串联四极杆质谱法进行分析[7~9],固相萃取法进行样品净化[9~11]。液相色谱串联四极杆质谱测定时,流动相溶剂、在线脱气机及色谱流路中均可能含有氟聚合物而引入背景干扰,部分组分如全氟己酸、全氟庚酸和全氟辛酸等化合物的仪器背景干扰值接近或高于定量限,难以实现PFASs的准确定量和有效监测;同时,水产加工食品的基质较鲜活水产品更为复杂,当样品中目标物残留较低时,采用四极杆质谱定性时部分化合物的两对MRM离子比率偏差较大,造成定性困难;另外,固相萃取净化方法的操作较繁琐,且对长碳链(C≥11)羧酸类PFASs的回收率不理想。针对上述问题,本研究结合杂质延迟技术,在液相系统混合器和进样器之间串联一根延迟色谱柱,去除了液相系统的背景值,并采用Qtrap质谱的多反应监测(MRM)信息依赖采集(IDA)增强子离子扫描(EPI)模式,实现了复杂基质中23种PFASs的同时定量定性分析,同时采用C18填料和石墨化碳黑(GCB)进行分散固相萃取净化,简化了样品前处理步骤,在保证全部目标物获得良好回收率的前提下,有效降低了复杂基质的干扰。这些改进显著提高了方法的准确性和可靠性,为准确监测水产加工食品中的PFASs污染水平提供了有效的技术保障。
2 实验部分
2.1 仪器、试剂与材料
Prominence UFLC液相色谱(Shimadzu公司);5500QTRAP四极杆线性离子阱复合质谱(AB SCIEX公司);T18 basic均质机(IKA公司);XW80A旋涡混合器(上海医大仪器厂);Himac CR 22GⅡ高速离心机(Hitachi公司);NEVAP 112氮吹仪(Organomation公司);MilliQ超纯水仪(Millipore公司)。
全氟丁酸、全氟戊酸、全氟己酸、全氟庚酸、全氟辛酸、全氟壬酸、全氟癸酸、全氟十一烷酸、全氟十二烷酸、全氟十三烷酸、全氟十四烷酸、全氟十六烷酸、全氟十八烷酸、全氟丁烷磺酸、全氟己烷磺酸、全氟庚烷磺酸、全氟辛烷磺酸、全氟癸烷磺酸、全氟辛烷磺酰胺、N甲基全氟辛烷磺酰胺、N乙基全氟辛烷磺酰胺、N甲基全氟辛基磺酰胺乙醇、N乙基全氟辛基磺酰胺乙醇、13C4全氟辛酸、13C4全氟辛烷磺酸、13C8全氟辛烷磺酰胺(Wellington Labortories公司);甲醇、乙腈(HPLC级,Merk公司);甲酸、乙酸铵(HPLC级,Fluka公司);超纯水(18.2 MΩ·cm); C18萃取剂(50 μm,艾杰尔公司);石墨化碳黑(CNWBOND CarbonGCB, 120~400目,CNW公司),其它未作特殊说明的试剂均为分析纯。
2.4 质量控制
为防止样品前处理步骤中引入高背景值,实验过程避免使用聚四氟乙烯材质的器皿,且器皿在使用前用超纯水和甲醇充分清洗。定量分析采用标准曲线校正,同位素内标法定量。每批样品进行测试时,同时做空白试验、基质加标试验和线性试验,空白试验确保试剂空白中所有分析物的含量低于定量限;基质加标试验加入PFASs标准物质(2 ng)和内标物(1 ng)验证方法的准确度,回收率控制在60%~120%范围内;采用至少5个浓度点进行标准曲线的绘制,且线性相关系数达0.99以上。
3 结果与讨论
3.1 杂质延迟法色谱条件的优化
依据色谱柱对液相系统中干扰物能捕集并释放的原理[11],流动相溶剂、在线脱气机和色谱流路中的PFASs在分析色谱柱中浓缩,可能与作为分析样品进样的目标物在同一时间检出。为了将来自液相系统的PFASs背景干扰组分与样品中的目标组分PFASs分离,在混合器和进样器之间安装延迟色谱柱(如图1),使干扰组分较目标组分延迟洗脱。
选用对PFASs有一定保留的色谱柱作为延迟柱[13],在液相系统混合器和进样器之间串联后,调整色谱的梯度洗脱条件, 使干扰组分在目标组分之后洗脱时基线分离(图2),达到了去除液相系统中背景干扰的目的。
3.2 质谱条件的优化
欧盟规定[14]用LCMS/MS方法检测有机污染物时,其定性确证方法要求每种化合物选用两对MRM离子,需要4个确证点(Identification Point,IP),即一个母离子,两个子离子(母离子IP为1.0,子离子IP为1.5)。但在本课题组前期建立的方法[7]中, PFBA, PFPeA, PFHxA, PFOSA, NMeFOSE和NEtFOSE仅能检测到一对稳定的MRM离子,且水产加工食品基质复杂,存在一定程度的基质抑制效应,当样品中目标物残留很低时,可能其它化合物的两对MRM离子也不能同时检测到,为定性分析带来极大困难。本方法首先采用目标物的单标溶液通过流动注射进质谱,以多反应监测负离子模式重点优化各化合物的子离子和碰撞能,增加了PFPeA, PFHxA和PFOSA的MRM扫描离子(见表1);然后利用MRMIDAEPI模式, 采用23种化合物的混合标准溶液通过自动进样器采集数据,该模式按照常规定量方法设定目标物的MRM离子对,在实际扫描过程中,尚某MRM通道采集的信号强度超过预设值后,就会触发EPI增强子离子扫描模式,进而获得对应MRM通道的母离子的增强二级离子全扫描质谱图,以用于定性分析; 同时建立目标物的在线EPI质谱库, 辅助定性。而此时,MRM通道采集的信号依然可以作为定量分析的数据。与MRM模式相比,EPI谱图由离子阱采集,其灵敏度高于四极杆,增强了二级碎片子离子扫描,且为全扫描质谱图,确证信息更为丰富,因而可以有效辅助痕量水平(例如≤0.1 μg/kg)或检出限浓度样品的定性确证,很好地解决了传统四极杆MRM模式对低浓度样品的定性难题。图3显示的是MRMIDAEPI模式与传统MRM模式下,0.1 μg/kg PFOA阳性沙丁鱼罐头样品的离子碎片信息。从图3可见,EPI扫描模式下,碎片离子响应增强近10倍,且该模式除采集母离子m/z 413和子离子m/z 369, 169外,还采集了m/z 219, 395等子离子的信息,确证点IP达7.0,远高于欧盟对有机污染物的确证要求,极大地提高了分析结果的可靠性。
3.3 样品前处理方法的优化
水产品加工食品的基质复杂,除脂肪、蛋白等内源性杂质外,还存在色素、糖等添加剂,本研究将鲜活水产品中PFASs的前处理方法[7]进一步优化,匀浆试样的取样量由5 g减为2 g,同时重点考察了净化填料用量的不同对目标物回收率和净化效果的影响。实验选取墨鱼丸样品为测试基质,按 2.2.1步骤进行提取,再添加2 ng PFASs标准物质,采用C18萃取剂吸附脂肪等非极性共萃物,通过对比50~250 mg C18对回收率和净化效果的影响,发现C18用量为150 mg时,平均回收率均优于其它用量(图4),且净化后的样品溶液澄清,去脂效果较好。在此基础上,添加GCB去除色素及具有共轭结构的谷氨酸、精氨酸等水产加工食品的风味成分,实验考察了150 mg C18与30~60 mg GCB的配比情况,GCB为50 mg时,可有效去除色素等杂质,且平均回收率高于90%(图5)。因此,本方法最终采用150 mg C18+50 mg GCB对样品提取液净化,在保证回收率的前提下,可有效降低水产加工食品中非极性和极性风味成分及脂肪酸等对测定的干扰。
3.4 灵敏度、准确度和精密度
取适量PFASs混合标准溶液和内标溶液,配制一系列浓度梯度的标准溶液,在2.3节条件下依次测定,以各组分和内标物的面积比值为纵坐标,各组分的质量浓度为横坐标进行线性回归分析。采用杂质延迟法,仪器的背景干扰得到了有效控制,5次平行测定中干扰组分的平均浓度不高于0.1 μg/L。在空白样品中添加低浓度的标准溶液,按2.2节步骤进行样品前处理后进样测定,以信噪比(S/N)≥10确定各组分定量限(LOQ),结果见表2。结果表明,23种化合物的线性良好,定量限为0.02~0.10 μg/kg,满足甚至高于目前食品基质中PFASs测定方法的指标[15,16]。
选取鱼丸、海米和沙丁鱼罐头为测试基质,分别添加相当于0.1, 1.0和10.0 μg/kg浓度的PFASs混合标准溶液进行测定,每个浓度做6个平行样品。同时做空白试验,扣除本底值后计算加标回收率和相对标准偏差。结果显示,PFASs在鱼丸中的加标回收率在72.2%~108.1%之间,相对标准偏差为5.2%~12.8%;PFASs在海米中的加标回收率在75.1%~116.4%之间,相对标准偏差为7.2%~13.5%;PFASs在沙丁鱼罐头中的加标回收率在67.5%~104.6%之间,相对标准偏差为5.9%~14.7%,方法的准确度与精密度良好。
3.5 实际样品分析
应用本方法分析了20份市售水产加工食品(鱼丸、虾丸、海米和沙丁鱼罐头各5份),发现部分样品存在PFASs污染(见表3),总量为0.82~7.437 μg/kg,并对阳性样品进行EPI谱库检索,匹配度均大于90%,且阳性样品中PFOA,PFOS和PFOSA的检出率较高,在样品中富集普遍。因此,建议开展水产加工食品中PFASs的残留监测,并以PFOA、PFOS和PFOSA为重点监控目标。
4 结 论
利用杂质延迟技术,通过在液相系统混合器和进样器之间安装延迟色谱柱,实现了色谱流路中PFASs假阳性干扰的去除。不过,由于延迟了目标物的保留时间,造成分析的平衡时间略有延长,但液相色谱系统的背景干扰得到有效控制,从而提高了方法的准确性;采用Qtrap质谱特有的MRMIDAEPI扫描模式实现一次进样同时定性与定量分析,有效判别假阳性样品,并解决痕量水平或检出限浓度样品的定性难题,提高了分析结果的可靠性;分散固相萃取的净化方式,简化了样品的前处理操作,降低了水产加工食品中风味成分及脂肪酸对测定的干扰。本方法的建立为进一步开展全国水产动物源性食品中全氟烷基化合物的残留水平调查提供了技术手段。
References
1 Wei Y H, Dai J Y, Liu M,Wang J S, Xu M Q, Zha J M, Wang Z J. Environ. Toxicol. Chem., 2007, 26(11): 2440-2447
2 Johansson N,Fredriksson A, Eriksson P. Neurotoxicology, 2008, 29(1): 160-169
3 Zheng L, Dong G H, Jin Y H, He Q C. Arch. Toxicol., 2009, 83: 679-689
4 WANG YaWei, CAI YaQi, JIANG GuiBin. Sci. Sin. Chim., 2012, 40(2): 99-123
王亚韡, 蔡亚岐, 江桂斌. 中国科学: 化学, 2012, 40(2): 99-123
5 Commission regulation (EC) No 2010/161/EU. Official Journal of the European Union, 2010
6 European Food Safety Authority.The EFSA Journal, 2011, 9(2): 2016
7 GUO MengMeng, WU HaiYan, LI ZhaoXin, TAN ZhiJun, ZHAI YuXiu. Chinese J. Anal. Chem., 2013, 41(9): 1322-1327
郭萌萌, 吴海燕, 李兆新, 谭志军, 翟毓秀. 分析化学, 2013, 41(9): 1322-1327
8 Wu Y N, Wang Y X, Li J G, Zhao Y F,Guo F F, Liu J Y, Cai Z W. Environ. Int., 2012, 42: 67-71
9 LAN Fang, FENG Sha, SHEN JinCan, WU XiaoPing, YUE ZhenFeng. Chinese J. Anal. Chem.,2013, 41(12): 1893-1898
蓝 芳, 冯 沙, 沈金灿, 吴晓萍, 岳振峰. 分析化学, 2013, 41(12): 1893-1898
10 Delinsky A D, Strynar M J, Nakayama S F, Varns J L, Ye X B, McCann P J, Lindstrom A B. Environ. Res., 2009, 109: 975-984
11 HE XiaoMin, CHEN Hao. Food Sci., 2014, 35(8): 193-197
贺小敏, 陈 浩. 食品科学, 2014, 35(8): 193-197
12 ZHANG XiuLan, GUO Jing, LI LingLing, DONG Liang, SHI ShuangXin, ZHANG LiFei, ZHOU Li, YANG WenLong, HUANG XingRu. Chinese J. Anal. Chem., 2014, 42(3): 452-456
张秀蓝, 郭 婧, 李玲玲, 董 亮, 史双昕, 张利飞, 周 丽, 杨文龙, 黄兴茹. 分析化学, 2014, 42(3): 452-456
13 Lacina O, Hradkova P, Pulkrabova J, Hajslova J. J. Chromatogr. A, 2011, 1218(28): 4312-4321
14 Commission Decision 2002/657/EC. Off. J. Eur. Commun., 2002, L221: 8
15 Hradkova P, Poustka J, louskova V, Pulkrabova J, Tomaniova M, Hajslova J. Czech. J. Food Sci., 2010, 28(4): 333-342
16 LI Jing, ZHANG Hong, CHAI ZhiFang, SHEN JinCan, YANG Bo. J. Instru. Anal., 2014, 33(10): 1109-1115
李 静, 张 鸿, 柴之芳, 沈金灿, 杨 波. 分析测试学报, 2014, 33(10): 1109-1115
act A comprehensive method for the simultaneous identification and detection of 23 perfluorinated alkyl substances (PFASs) in processed aquatic products by impurity delay was developed using liquid chromatography coupled with quadrupole/linear ion trap tandem mass spectrometry (LCQ/TrapMS). The sample was extracted with acidified acetonitrile, cleanedup by dispersive solid phase extraction using C18 and graphitized carbon blacks (GCB). The separation of 23 PFASs was performed on a Kinetex XBC18 (100 mm×2.1 mm,2.6 μm) column using gradient elution of 5 mmol/L ammonium acetate and methanol as mobile phase. And a short C18 HPLC column was inserted between the mixer and the autosample, which delayed compounds coming from the LC system. A scheduled multiple reaction monitoring (MRM) in negative mode as survey scan and an enhanced product ion (EPI) scan as dependent scan in an informationdependent acquisition (IDA) experiment were adopted in mass spectrometry acquisition. Online labbuilt MS/MS library and the isotope internal standards were employed for the identification and quantification. The calibration curves for the detection of 23 PFASs were linear well with correlation coefficient over 0.995. The limits of quantification for all analytes were ranged from 0.02 μg/kg to 0.1 μg/kg. The average spiked recoveries for 23 PFASs were between 67.5% and 116.4%, with relative standard deviations (RSDs) from 5.2% to 14.7%. The background coming from the part of LC was controlled well by the impurity delay. The proposed method can be used to identify and detect the 23 PFASs in a single run, and also suitable for the analysis of processed aquatic product samples.
Keywords Perfluorinated alkyl substances; Processed aquatic products; Impurity delay; Liquid chromatographyquadrupole/linear ion trap tandem mass spectrometry
(Received 5 February 2015; accepted 12 June 2015)
This work was supported by the Special Foundation for Basic Research Program of the Ministry of Science and Technology, China (No.20144FY30100)
