二氢色原酮-3-甲醛的合成工艺研究
王晓丽 严丽 褚朝森
摘 ?????要:以邻羟基苯乙酮和三氯氧磷为原料,N,N-二甲基甲酰胺为溶剂,合成二氢色原酮-3-甲醛,总收率93%,纯度99.7%,可生产1.19 kg/批。通过研究放大实验反应溶剂用量、反应时间和析晶时间对产物收率的影响,确定了最佳反应条件为N,N-二甲基甲酰胺5 L,反应24 h,析晶1.5 h。室温下,用甲醇和乙酸乙酯混合溶剂打浆纯化代替重结晶,提升产物纯度,简化了操作,降低了成本。
关 ?键 ?词:二氢色原酮-3-甲醛;三氯氧磷;N,N-二甲基甲酰胺
中图分类号:TQ215 ??????文献标识码: A ???????文章编号: 1671-0460(2019)03-0501-04
Abstract: Using 2'-hydroxyacetophenone and phosphorus oxychloride as starting materials, DMF as solvent, chromone-3-carboxaldehyde was synthesized, and the yield was 93%,the purity was 99.7%,the output per batch was 1.19 kg. The effect of solvent dosage, reaction time and crystallization time on the yield was studied. The best reaction conditions were determined as follows: DMF 5 L, the reaction time 24 h,the crystallization time 1.5 h. At room temperature, mashing with methanol and ethyl acetate instead of recrystallization can improve product purity significantly. The operation was simplified with low costing.
Key words: Chromone-3-carboxaldehyde; Phosphorus oxychloride; DMF
二氢色原酮-3-甲醛,又名色酮-3-甲醛,其分子中含有醛基、羰基和C-C雙键,容易发生加成、取代等多种化学反应[1],可用于合成多种生理活性化合物[2],例如抗癌、抗炎、抗氧化、抗高血压、抗病毒药物[3]等。
目前二氢色原酮-3-甲醛的合成方法已有报道。Xiao Jiang等[4]以3-溴色酮为原料,利用醋酸钯催化剂合成目标产物,该方法需要惰性气体保护,且原料价格昂贵,不适合放大生产;Xiang Haoyue[5],Sharma[6]等以2-羟基苯乙酮为原料,室温下合成了目标产物,操作较为简便,然而2-羟基苯乙酮价格昂贵,不经济;Rajanna等[7]首次采用邻羟基苯乙酮为原料,在N,N-二甲基甲酰胺(DMF)存在下和三氯氧磷反应制备目标产物,由于该方法原料便宜,操作简便,备受学者关注[8-10]。该方法的研究存在以下问题:收率偏低(47%~87%不等),若想提高收率需要惰性气体保护;目前的研究仅达到克级;为提高反应效率需要高温。随着二氢色原酮-3-甲醛的市场需求量不断增加,这些问题均限制了其工业化发展。本研究以此为切入点,选择邻羟基苯乙酮与三氯氧磷的反应作为研究对象,优化反应条件,实现了公斤级生产,为二氢色原酮-3-甲醛的量化生产提供参考数据。
1 ?实验方法
1.1 ?实验材料
邻羟基苯乙酮为工业级(纯度98%),购自江苏海翔化工有限公司;三氯氧磷为工业级(纯度98%),购自淮安华源化工有限公司;其余试剂均为市售工业级试剂。
1HNMR在德国Bruker公司AM500MHz共振仪上测定;熔点在上海精密科学仪器有限公司WRS-B型熔点仪上测定。纯度在日本岛津高效液相色谱仪(LC-2010型)上测定。
1.2 ?实验方法
1.2.1 ?小试实验
参照文献[11]方法,取200 g邻羟基苯乙酮于2 L的三口圆底烧瓶中,加入DMF880 mL,冰浴下搅拌,滴加三氯氧磷368 mL(滴加过程保持反应液內温小于10 ℃),滴完后自然升温至室温,继续搅拌至反应完全,薄层色谱(TLC)检测反应进程。反应结束后,将反应液倒入1.6 L冰水中,搅拌0.5 h,过滤得黄色固体。收集固体,甲醇500 mL重结晶得黄白色固体,30 ℃下真空干燥12 h得黄白色固体粉末230 g,收率90%。熔点151~152.3 ℃;1HNMR(CDCl3):δ=7.53(m,2H,Ar-H);7.76(m,1H,Ar-H);8.31(t,1H,CH=);8.57(s,1H,Ar-H);10.41(s,1H,CHO)。
1.2.2 ?放大实验
以小试实验为基准进行放大,取1 000 g邻羟基苯乙酮于10 L四口圆底烧瓶中,加入DMF 5 L,冰浴下搅拌,滴加三氯氧磷1.84 L,滴完后自然升温至室温,继续搅拌反应24 h,反应完全。将反应液倒入10 L冰水中,搅拌1.5 h,过滤得黄色固体粗品,收率95%。收集固体,加入1 L甲醇和1 L乙酸乙酯搅拌0.5 h,过滤,收集滤饼,30 ℃下真空干燥12 h得黄白色固体粉末1 190 g,收率93%。核磁检测正确。
1.2.3 ?高效液相色谱(HPLC)检测产物纯度
产物纯度HPLC检测采用C18色谱柱,流动相为75%乙腈水溶液,检测波长为254 nm,流速为1 mL/min。
2 ?结果与讨论
小试实验与文献[11]数据基本吻合,反应条件亦参照文献条件进行。实验过程中三氯氧磷宜采用慢速滴加的方式加入,这是因为反应过程中放热明显,过快的滴加速度会导致放热剧烈,瞬间产生大量的氯化氢气体,存在安全隐患。通过小试实验发现溶剂的用量、反应时间、搅拌析晶时间是影响产物收率和质量的关键因素,从工业化的角度出发,本着保证质量和降低成本的原则,参考文献方法[12],考查了放大阶段的反应溶剂用量、反应时间和搅拌析晶时间对产物收率的影响,并对终产物的纯化方法进行了优化。
2.1 ?溶剂的用量对反应的影响
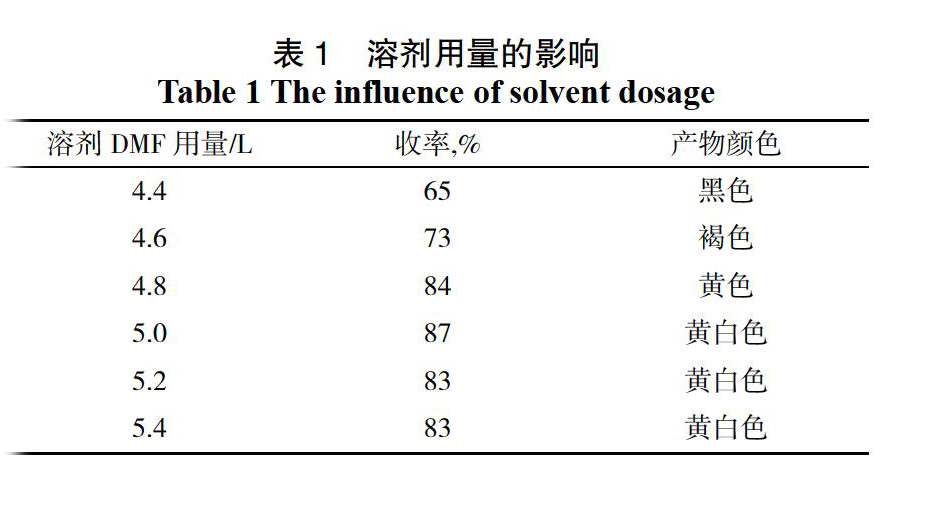
用邻羟基苯乙酮1 000 g,三氯氧磷1.84 L,室温下反应20 h,搅拌析晶1 h,产物未做纯化处理,考查溶剂DMF不同用量对反应效果的影响,实验结果见表1。
实验结果表明,溶剂DMF用量不同时产物收率差异较大,同时产物的颜色也出现了变化。当DMF用量为4.4 L和4.6 L时,产物的收率较低(低于73%),这是由于反应液浓度偏高,粘度大,搅拌不充分;该反应为放热反应,反应液粘度大导致热量无法散发,高温氧化致使产物颜色较深。DMF用量超过4.8 L时,收率出现了明显的上升,同时产物颜色变淡(黄色或黄白色),其中DMF用量为5.0 L时收率最高(87%),用量大于5.0 L时收率有所下降,这是因为溶剂用量偏大導致反应体系变烯,分子间接触的概率下降。综合考虑,溶剂DMF用量为5.0 L时反应效果最佳。
2.2 ?反应时间对收率的影响
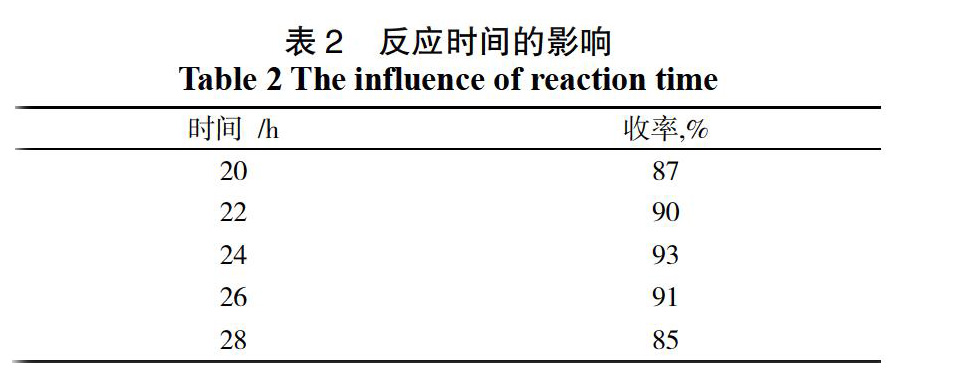
用邻羟基苯乙酮1 000 g,三氯氧磷1.84 L,DMF 5 L,室温下反应,搅拌析晶1 h,产物未做纯化处理,以2 h为一个区间,考查不同时间对反应效果的影响,结果见表2。
实验结果表明,反应收率随着时间的延长出现了变化,反应由20 h延长至24 h,收率逐步升高,最高达93%,继续反应,收率则出现下降趋势,反应至28 h,收率降低至85%,证明该反应体系稳定,产物醛有氧化现象导致收率降低。综合考虑,反应24 h最佳。
2.3 ?析晶时间对收率的影响
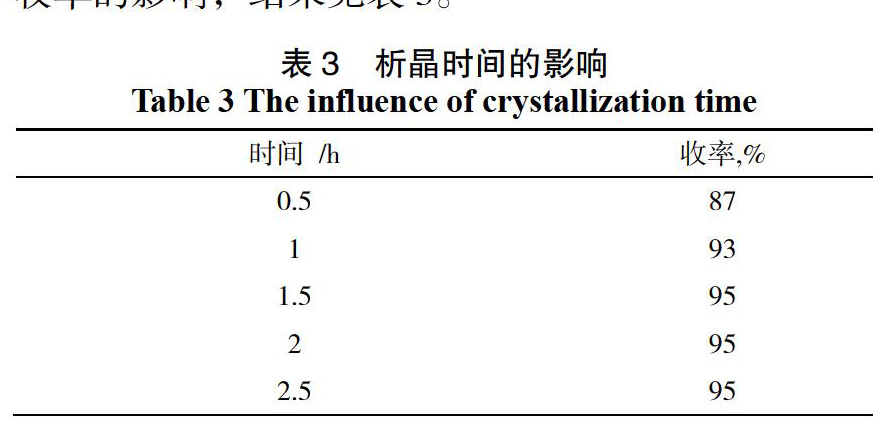
用邻羟基苯乙酮1 000 g,三氯氧磷1.84 L,DMF 5 L室温下反应24 h,将反应液倒入10 L冰水中搅拌,产物未做纯化处理,考查不同析晶时间对产物收率的影响,结果见表3。
实验结果表明,搅拌析晶时间长有利于产物的析出,当搅拌时间超过1.5 h后,产物收率达到平衡(95%),证明产物已完全析出,之后继续搅拌,未出现产率下降的现象,说明冰水中搅拌析晶,产物较稳定,综合考虑,最佳搅拌析晶时间为1.5 h。
研究证明,二氢色原酮-3-甲醛的最佳反应条件为邻羟基苯乙酮1 000 g,三氯氧磷1.84 L,DMF 5 L,室温下反应24 h,反应液倒入10 L冰水中搅拌1.5 h,粗产物收率最高达到95%。
2.4 ?产物的纯化
小试阶段产物的纯化可以参照文献方法进行重结晶,放大后,产物重结晶用甲醇量较大(达到2.5~3 L),且重结晶过程需要较长的时间,从节约成本、提高收率的角度考虑,实验尝试了几种混合溶剂对粗产物进行打浆纯化,实验结果见表4。
结果表明,不同种类的混合溶剂打浆纯化效果存在的明显差异。由于二氯甲烷对产物的溶解性较好,使得含有二氯甲烷的混合溶剂纯化收率总体较低;石油醚对产物的溶解性最差,表现为含有石油醚的混合溶剂纯化收率较高,但产物纯度偏低;总体看来甲醇和乙酸乙酯混合溶剂打浆效果最好,收率为93%,产物纯度达99.7%。
为进一步研究甲醇和乙酸乙酯在打浆纯化中的作用,实验用1 L甲醇打浆后过滤,干燥、检测,发现产物中仍部分极性较小的杂质无法去除,此时产物纯度仅有94.5%(HPLC),说明甲醇可以去除粗品中的大极性杂质。将甲醇处理后的产品用1 L乙酸乙酯再次打浆,过滤,滤饼采用1 L乙酸乙酯洗涤后,干燥、检测,产物纯度提高至99.7%(HPLC),符合市售标准。用甲醇和乙酸乙酯混合溶剂打浆纯化代替重结晶,提高了产品收率和纯度,降低了劳动的强度和时间,降低了工业化成本。
综上所述,二氢色原酮-3-甲醛的最佳反应条件为邻羟基苯乙酮1 000 g,三氯氧磷1.84 L,DMF 5 L,室温下反应24 h,反应液倒入10 L冰水中搅拌1.5 h,粗产物收率最高达到95%。用甲醇和乙酸乙酯混合溶剂打浆纯化代替重结晶,产物收率为93%,纯度达99.7%。
2.5 ?产物纯度的检测
将待测样品配制成0.5 mg/mL的乙腈溶液,在254 nm波长下,以75%乙腈水溶液为流动相,检测产物纯度,结果见图1。
结果显示,产物出峰时间为2.77 min,杂质出峰时间为3.30 min,产物纯度99.7%。实验尝试以甲醇代替乙腈,以期降低成本,提高安全性,但甲醇的洗脱性能较差,峰型不符合要求,因此确定乙腈为最佳流动相。
2.6 ?二氢色原酮-3-甲醛衍生物的制备
在优化反应条件的基础上,实验尝试合成了几种二氢色原酮-3-甲醛的衍生物,结果见表5。
结果表明,利用实验获得的最佳工艺合成二氢色原酮-3-甲醛的衍生物获得了成功,产物纯度均大于98%,符合市售标准,其中6-溴色酮-3-甲醛和6-氯色酮-3-甲醛的收率在90%以上。证实本研究所建立的优化工艺条件具有普遍适用性。
3 ?结 论
以邻羟基苯乙酮和三氯氧磷为原料,成功制备了二氢色原酮-3-甲醛,每批可生产1.19 kg。通过研究放大实验阶段反应溶剂用量、反应时间和搅拌析晶时间对产物收率的影响,确定了最佳反应条件,粗产物收率最高达到95%。优化了产物纯化方法,用甲醇和乙酸乙酯混合溶剂打浆纯化代替重结晶,产物最终收率为93%,纯度达99.7%,此法简化了操作,降低了劳动的强度和时间,降低了工业化成本。
参考文献:
[1]W. K. Sua, Z. H. Lia, L. Y. Zhao. One-Pot Sythesis of 3-formylchromones from bis-(trichloromethyl) Carbonate/DMF[J]. Organic Preparations and Proceduresint., 2007,39 (5), 495-502
[2]B. China Raju, R. Nageswara Rao, P. Suman, et al. Synthesis, structure–activity relationship of novel substituted 4H-chromen -1,2,3,4-tetrahydropyrimidine-5-carboxylates as potential anti- mycobacterial and anticancer agents[J]. Bioorganic & Medicinal Chemistry Letters, 2011, (21): 2855–2859.
[3] G. Mahaboob Basha, S. Kumar Yadav, R. Srinuvasarao, et al. A mild and efficient protocol to synthesize chromones, isoflavones, and homoisoflavones using the complex 2,4,6-trichloro- 1,3,5- triazine/ dimethylformamide[J]. Can. J. Chem., 2013 (91): 763–768.
[4] Xiao Jiang, Jin-Mei Wang, Ying Zhang, et al. Palladium- Catalyzed Formylation of Aryl Halides with tert-Butyl Isocyanide [J]. Organic Letters, 2014, 16(13): 3492-3495.
[5] Xiang Haoyue, Qi Xue yu, Xie Yuyuan, et al. One-pot syntheses of novel pyrazole-containing bisphosphonate esters at room temperature [J]. Organic and Biomolecular Chemistry, 2012, 10(38): 7730-7738.
[6] Sharma, Vinay Prabha, Kumar, Praveen. Synthesis, Spectral Studies and Antibacterial Activity of 3-(4-Phenyl-2, 3-dihydro-1,5– benzodiazepin -2-yl)chromone[J]. Asian Journal of Chemistry, 2014, 26(13): 3992-3994.
[7] Rajanna, Solomon, Florence, Moazzam Ali, Mir, et al. Kinetics and mechanism of Vilsmeier-Haack synthesis of 3-formyl chromones derived from o-hydroxy aryl alkyl ketones: A structure reactivity study [J]. Tetrahedron, 1996, 52(10): 3669- 3682.
[8] Nikitina, Polina A., Kuz'Mina, Ludmila G., Perevalov, Valery P., et al. Synthesis and study of prototropic tautomerism of 2- (3- chromenyl) -1-hydroxyimidazolesOriginal Research Article[J]. Tetrahedron, 2013, 69(15): 3249-3256.
[9] Zwergel, Clemens, Valente, Sergio, Salvato, Angela, et al. Novel benzofuran–chromone and –coumarin derivatives: synthesis and biological activity in K562 human leukemia cells[J]. MedChemComm, 2013, 4(12) 12: 1571 – 1579.
[10]Zhu, Wufu, Chen, Chen, Sun, Chengyu, et al. Design, synthesis and docking studies of novel thienopyrimidine derivatives bearing chromone moiety as mTOR/PI3Kα inhibitors[J]. European Journal of Medicinal Chemistry, 2015, 93(26): 64-73.
[11]Abou-Melha, Khlood S. Octahedral Co(Ⅱ) and Ni(Ⅱ) complexes of Schiff bases, semicarbazone and thiosemicarbazone, synthesis, biological, spectral, and thermal studies[J]. Journal of Coordination Chemistry, 2008, 61(13):2053-2067.
[12]董乙文,嚴丽,王晓丽,等. 3-(2-噻吩)丙烯酸的合成工艺研究[J]. 当代化工,2016,45(11):2517-2519.
