利培酮和帕利哌酮抗精神病活性的理论研究
祖国平 郭琴 王亚丽

摘 ?????要:采用密度泛函理论和时间分辨密度泛函理论,优化利培酮和帕利哌酮的稳态构型,计算Wiberg键级、紫外-可见分子吸收光谱、分子轨道、Hirshfeld原子电荷、福井函数等,预测利培酮和帕利哌酮的活性点位,解释其药理学行为的化学基础。结果表明帕利哌酮上引入的端羟基可降低临近原子的反应活性,从而减轻其药物副作用。
关 ?键 ?词:利培酮;帕利哌酮;密度泛函理论;药物活性
中图分类号:O641.12 ??????文献标识码: A ??????文章编号:1671-0460(2019)01-0017-03
Abstract: With the help of DFT and Time-Dependent DFT, the stable structures of Risperidone and Paliperidone were optimized, and then Wiberg bond order, UV-Vis spectra, frontier molecular orbital, atomic charge, etc., were calculated, which can predict the active sites and explain the difference of pharmacological behaviors. The results indicated that the terminal hydroxyl group on Paliperidone can greatly affect the reactivity of adjacent atoms, which can decrease its pharmaceutical side-effect.
Keywords: Risperidone; Paliperidone; Density functional theory (DFT); Pharmaceutical activity
精神分裂症是一种慢性疾病,目前,其治疗通常采取药物联合心理干预的方式[1,2]。利培酮是一种治疗精神分裂症的非典型抗精神病药,其疗效显著,但是可能导致锥体外系反应等副作用,由此影响其临床应用。而帕利哌酮,即9-羟利培酮,其疗效与利培酮相当,却克服了利培酮的不良作用,因而渐渐被患者接受[3,4]。
利培酮和帕利哌酮都属于苯丙异噁唑类衍生物,其确切药理机制尚不清楚,一般认为是通过拮抗多巴胺D2和5-羟色胺2A(5HT2A)受体发挥抗精神病作用[5,6]。因此,深入研究二者与各种神经递质受体的结合在其疗效及各种副反应中的确切作用,不但能揭示药物产生疗效和各种副作用的机理,还可能对精神疾病的产生原因有进一步的认识。由于实验研究难以揭示药物药理作用的详细信息,而量子化学方法可以在分子层面预测药物与受体的结合方式,研究药物的作用机理和可能的副产物,从而为药物筛选提供理论依据[7,8]。
利用密度泛函理论和时间分辨密度泛函理论,得到了利培酮和帕利哌酮的稳态构型、吸收光谱、电子排布、福井函数等信息,对其可能的作用方式进行初步研究。目前,国内外对抗精神病药物分子的理论研究非常少见,我们对该领域进行了一些积极的探索。
1 ?实验部分
本文选用利培酮和帕利哌酮两种典型药物为研究对象,运用密度泛函理论(Density Functional Theory,DFT)和时间分辨密度泛函理论(Time- Dependent DFT,TD-DFT),在B3LYP/6- 31G**水平下,选取溶剂极化连续模型,采用Gaussian 09 D01软件计算。分子轨道由GaussView 6.0绘制,吸收光谱由GaussSum 2.2绘制,原子电荷则采用Multiwfn 3.4计算。
2 ?结果与讨论
2.1 ?分子结构
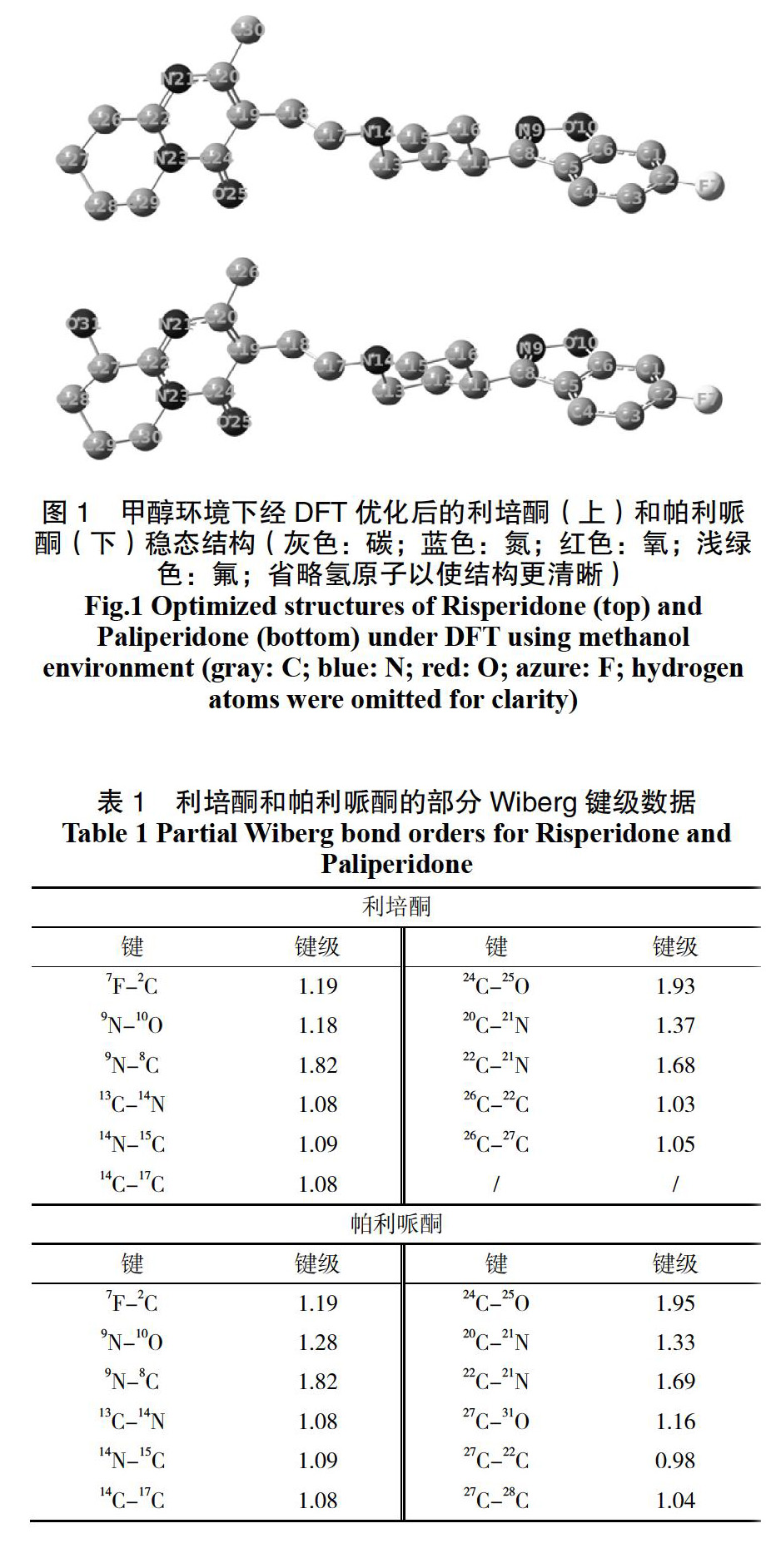
利培酮和帕利哌酮的基态分子结构分别在甲醇、正己烷和水三种溶剂环境中进行优化计算。三种溶剂环境中的优化结果很类似,仅在键角方面有细微差别,误差不超过1%,可见这两种药物分子均有很好的溶剂稳定性。图1所示是甲醇条件下的优化结果。利培酮和帕利哌酮的分子结构很类似,且优化后的结构骨架几乎保持一致,仅仅存在一个端羟基的差异。可以预见,该端羟基可能导致二者不同的药理作用。
进一步研究了两种药物分子的Wiberg键级[9],见表1。键级是相邻原子间成键强度的量度。一般而言,键级越大,成键越稳定。帕利哌酮上引入羟基使临近单键的键级增大,成键稳定性提高,因此该点位的化学反应活性降低,从而减少副反应的发生。端羟基的引入對更远处的键级影响很小,这是因为该体系的共轭性不高,不利于电子离域。
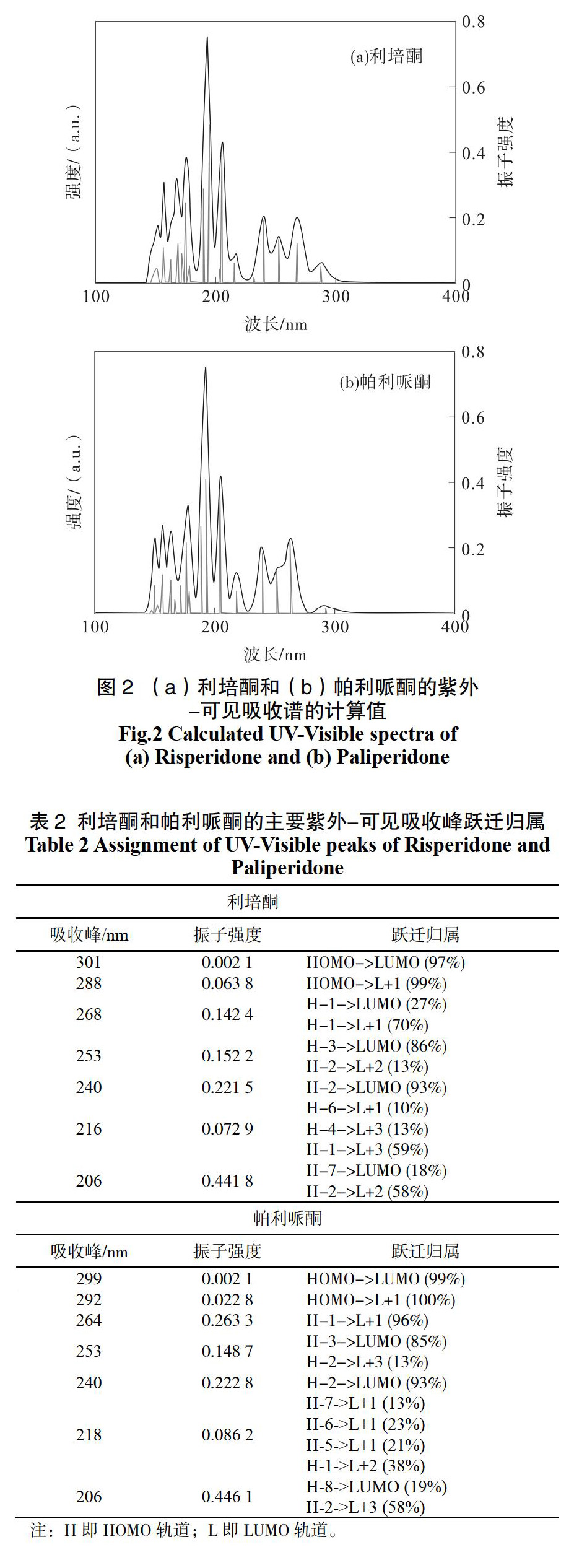
量化计算可以描述紫外-可见吸收光谱的本质特征。图2是利培酮和帕利哌酮的吸收光谱的理论值。利培酮和帕利哌酮HOMO-LUMO跃迁位于均位于波长λ=300 nm处,但是其振子强度太小,几乎观察不到。由表2可见,对于性质一致的跃迁类型,帕利哌酮比利培酮的吸收谱峰蓝移约3~10 nm,这可以归因于端羟基的引入略微增加了体系的扭曲程度,降低了分子共轭度,使电子跃迁所需克服的能垒升高了。可以预见利培酮活性更高,可能会有不良副反应。
2.3 ?反应活性
2.3.1 ?分子轨道
利培酮和帕利哌酮的分子轨道如图3所示。通常200 nm左右以下的吸收峰可归属为来源于内层轨道跃迁,不在本文讨论范围内,故该部分主要讨论240 nm及以上的吸收峰。对于利培酮和帕利哌酮,240 nm和253 nm的跃迁的位置和振子强度几乎完全一致,且都可归属为HOMO-3到LUMO和HOMO-2到LUMO的跃迁,即从苯丙异噁唑基团到苯并嘧啶基团的跃迁。不同的是,帕利哌酮在264 nm展现出HOMO-1到LUMO+1的强吸收峰,而利培酮的HOMO-1到LUMO+1的吸收峰较弱,且红移至268纳米。从图3可以看出,HOMO-1和LUMO+1都位于苯并嘧啶基团,且部分弥散至端羟基,由此可见端羟基对吸收光谱的明显影响。
2.3.2 ?原子电荷
本文采用变形密度分割的Hirshfeld方法[10,11]计算原子电荷,结果见表3。在远离端羟基的位置,利培酮和帕利哌酮的原子电荷几乎一致。在端羟基附近,帕利哌酮上的31O使原来差异明显的原子电荷趋向于平均,增加了体系的稳定性,从而降低了反应活性。
2.3.3 ?福井(Fukui)函数
我们进一步计算了利培酮和帕利哌酮的Fukui函数[12,13]。在图4(b)和(d)中,亲核进攻因子?+1显示负值绿色区域的反键轨道,表明此处是亲核试剂最易进攻的部位。利培酮和帕利哌酮差别不大,表明二者受亲核试剂进攻的概率类似。而在图4(a)中,利培酮的亲电进攻因子?-1显示19C,20C,21N,22C等原子的成键轨道为正值蓝色区域,是亲电试剂最易进攻的部位;而在图4(c)中,帕利哌酮的亲电进攻因子?-1显示19C,20C,21N,22C等原子几乎没有成键轨道的正值蓝色区域。二者的差异可归因于端羟基的引入。随着原子距离的增加,由于该体系的共轭性被破坏,18C,17C,15C,14N,13C和12C等原子受端羟基的影响几乎可以忽略不计,此时利培酮和帕利哌酮展现出类似的亲电进攻因子。此外,利培酮的25O也显示出更大的成键轨道。由此可知利培酮具易受亲电试剂进攻,可以从一定程度上解释其易造成锥体外系反应的原因。
3 ?结 论
本论文利用密度泛函理论优化利培酮和帕利哌酮的稳态构型,计算Wiberg键级、紫外-可见分子吸收光谱、分子轨道、Hirshfeld原子电荷、福井函数等,证明帕利哌酮上端羟基的引入降低了临近原子的反应活性,从而抑制了副反应的发生。目前,国内外对抗精神病药物分子的理论研究非常少见,我们对该领域进行了一些积极的探索。
参考文献:
[1]毛丽. 利培酮对青少年精神分裂症患者认知功能影响[J]. 精神医学杂志, 2016, 29 (5): 369-371.
[2]贺楚梅, 阳前军, 王珍兰. 帕利哌酮与利培酮治疗精神分裂症的临床观察[J]. 中国药房, 2016, 27 (11): 1542-1544.
[3]乔屹, 梅轶, 盛建华. 多巴胺D2受体基因Taq1A多态性与利培酮、帕利哌酮所致高泌乳素血症的关系[J]. 临床精神医学杂志, 2016 , 26 (6): 399-401.
[4] 孙云峰, 应茵, 夏仲尼. 帕利哌酮与利培酮治疗精神分裂症的疗效和安全性Meta分析[J]. 中国现代应用药学. 2014 , 31 (10): 1263-1267.
[5]李良, 李建华, 杨剑虹, 等. 帕利哌酮与利培酮对首发男性精神分裂症患者糖脂代谢影响的对照研究[J]. 中国药学杂志, 2013, 48 (8): 649-651.
[6]王来海, 张瑞岭,张红星,等. 帕利哌酮与利培酮治疗首发精神分裂症的临床疗效与安全性[J].中国医院用药评价与分析,2012,12(8): 725-728.
[7]郭琴, 祖国平, 王亚丽, 等. 基于密度泛函理论的奥氮平和氯氮平光谱性质和药物活性研究[J]. 河南科学, 2017, 35 (11): 1755-1761.
[8]N. Li, P. Quan, X. Wan, C. Liu, X. Liu, L. Fang. Mechanistic insights of the enhancement effect of sorbitan monooleate on olanzapine transdermal patch both in release and percutaneous absorption processes[J]. European Journal of Pharmaceutical Sciences, 2017, 107: 138-147.
[9]K. B. Wiberg. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane[J]. Tetrahedron, 1968, 24 (3): 1083-1096.
[10]F. L. Hirshfeld. Bonded-atom fragments for describing molecular charge densities[J]. Theoretical Chemistry Accounts, 1977, 44 (2), 129-138.
[11]徐洋,羅海霞,范强, 等. 新药萘夫西林药理性质的密度泛函研究[J]. 当代化工, 2015, 44 (4): 699-701.
[12]R. G. Parr, W. Yang. Density functional approach to the frontier-electron theory of chemical reactivity[J]. Journal of the American Chemical Society, 1984, 106(14): 4049-4050.
[13] 陈莹, 徐抗震, 宋纪蓉, 等. 酚酸抗氧化活性的理论计算[J]. 食品科学, 2011, 32 (9): 36-39.
