肟醚类化合物作为导向基团在钯催化C-H活化中的应用
程肖丁
摘 ?????要: 肟醚结构中氮原子可与过渡金属配位成金属螯合物,诱导整个反应体系的定向和化学选择性催化。目前,肟醚衍生物作为导向基团广泛应用于过渡金属催化的C-H官能化,构建C-C键、C-N键、C-X键、C-O键等。肟醚除可诱导sp2/sp3 C-H活化,实现C-H键的官能化外,还可被脱去生成酮醇类,与过渡金属合成稳定配合物,亦可作为氧化剂参与反应。介绍了肟醚类化合物作为导向基团在钯催化C-H活化领域中的应用和进展。
关 ?键 ?词:肟醚;C-H活化;钯催化;导向基团
中图分类号:TQ 032 ??????文献标识码: A ??????文章编号: 1671-0460(2019)01-0092-06
Abstract: Oxime ethers can coordinate with transition metals to form the metal chelates,which can induce the directional and chemoselective catalysis of the entire reaction system. As directing groups, oxime ethers are widely used in C-H functionalization catalyzed by transition metals, constructing C-C bonds, C-N bonds, C-X bonds, C-O bonds,C-N bonds and so on. In this paper, the application and research progress of oxime ethers as directing groups in the palladium-catalyzed C-H activation were described. It's pointed out that oxime ethers can be used to direct sp2/sp3 C-H activation.
Key words: Oxime ethers; C-H activation; Pd-catalysis; Directing groups (DG)
一直以来,肟醚类化合物在有机化学以及植物化学领域中占据着重要地位。肟和肟醚的基本结构单元(C=NO-R)是很多药物的关键骨架,具有抗菌、抗肿瘤和消炎等生物活性,被广泛应用于医药领域[1-3]。此外,肟醚类化合物还是多功能的化学中间体和前体,可通过化学反应如亲电加成、环化加成、过渡金属催化交叉偶联等生成一系列含氮化合物[4-6]。
由于肟醚结构中氮原子孤对电子的存在,使得该结构呈现出一定的路易斯碱特性,因此可与过渡金属配位络合,是一些过渡金属催化的有机转化中重要的诱导基团,尤其是在钯催化的C-H键的活化方面。
C-H键活化是构建C-C键的重要策略之一,最早可以追溯到19世纪末人们发现的金属与碳氢化合物之间的的反应,然而真正实现过渡金属参与催化的C-H键活化是在20世纪60年代,以Fujiwara[7]的烯烃芳基化为典型代表。一般地,底物的C-H键因其所处位置不同、取代基不同而导致带电性不同,因此,对C-H键进行选择性的官能团化是可以控制并实现的。对于某些底物,可以通过路易斯碱诱导基团对金属催化剂配位,从而控制分子内的碳氢活化。常见的导向基团有烷基甲酰基、烷基甲酰胺基、烷基甲酰氧基、羧基、2-吡啶基、亚胺、肟醚、恶唑啉以及N-甲氧基胺基甲酰基等。目前,关于肟醚作为导向基团在C-H活化方面的应用已成为C-H官能化中的重要研究领域和方向。
本文根据近几年来肟醚作为导向基团在碳氢活化方面的应用,系统地概述肟醚的诱导作用并作出自己的见解和展望,主要从肟醚参与诱导的C-H官能化类型以及产物的延伸方面做出详细阐述。
1 ?C-H键的官能化
1.1 ?C-H键的乙酰氧基化
目前,关于sp3 C-H键的常规氧化催化方法已经在合成化学中广泛应用。然而,由于sp3 C-H 键的键能较大,且氧化产物易于被氧化剂过度氧化以及在某些复杂有机分子的存在下难以实现区域选择性官能化,所以sp3 C-H 键的催化氧化仍然具有挑战性。
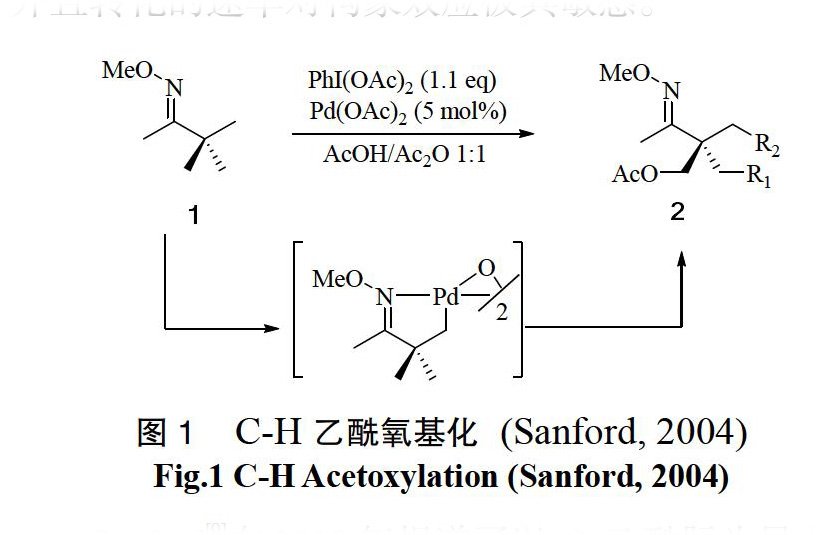
甲烷和一些更复杂烷烃的C-H键的催化氧化条件往往比较苛刻,且底物耐受性较差。2004年,Sanford[8]报道了一种选择性钯催化氧化芳烃和苄型C-H键的新方法。他发现在含有肟基团和吡啶骨架的底物中,未取代的sp3 C-H键可在氧化剂PhI(OAc)2存在下进行高区域化学选择性的Pd(II)催化。该催化同时具备反应可行性和空间诱导选择性,Pd(II)与底物生成的螯合物诱导和促进sp3 C-H键的激活,同时,螯合物依据烷烃底物的空间和电子性质定向活化C-H键,实现C-O键的构建。
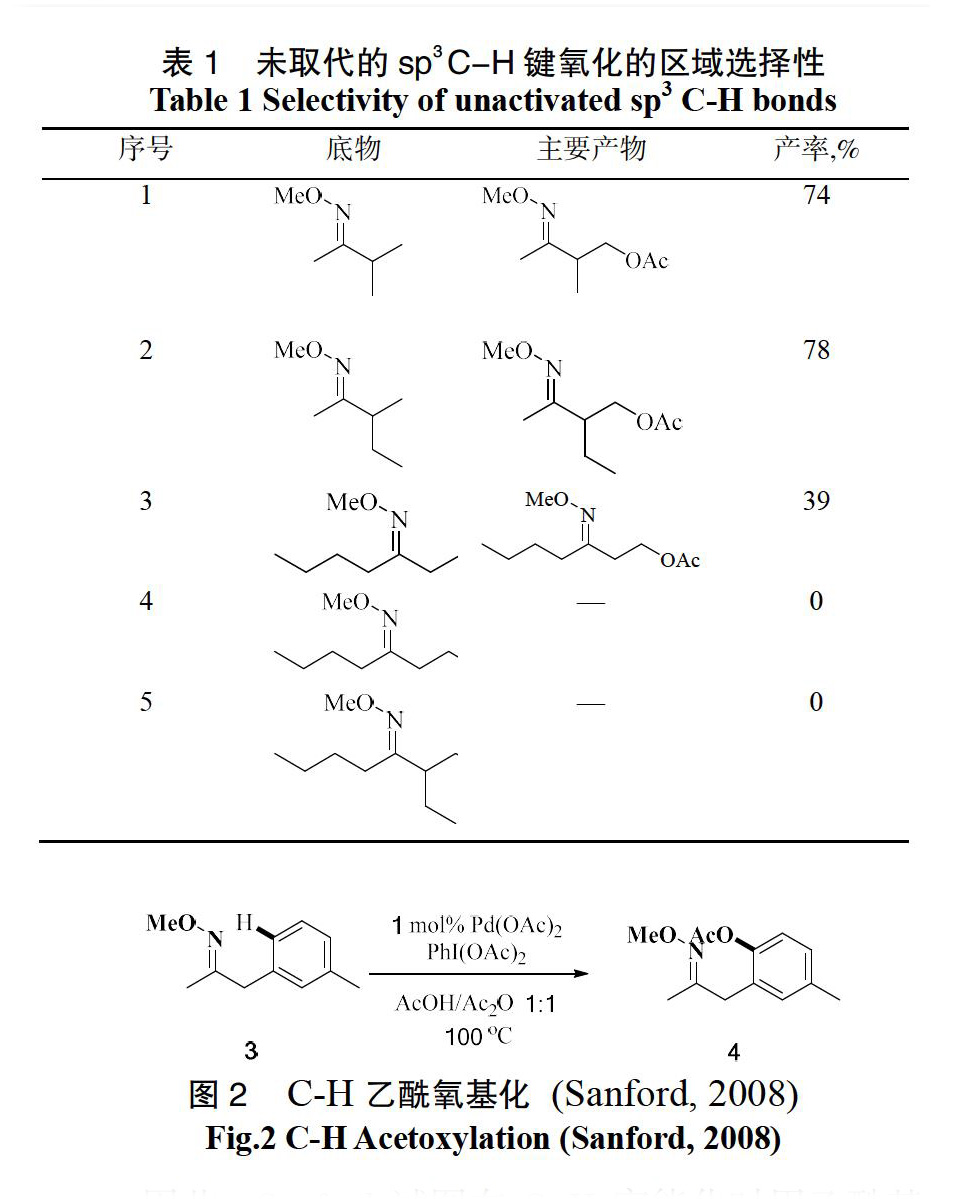
Sanford研究的烷烃催化氧化主要集中在频哪醇酮O-甲基肟的官能化,在催化剂Pd(OAc)2 (5 mol%)和氧化剂PdI(OAc)2 (1.1 eq)作用下得到了β-含氧产物(图1)。随后,Sanford在相同的反应条件下,探索底物适用性范围及转化的区域选择性。实验得出,一系列O-甲基肟底物分别以0%~%的产率得到相应的β-含氧产物(表1)。此外,Sanford发现取代环酮肟也是良好的氧化底物和诱导基团,并且转化的速率对构象效应极其敏感。
Sanford[9]在2010年报道了以O-乙酰肟为导向基团的Pd(II)催化的sp2和sp3 C-H官能化反应,且C-H官能化产物可转化为邻位或者β-官能化的酮、醇、胺和杂环。酮类化合物是有机合成中通用且应用广泛的合成中间体。然而,酮是Pd(II)的不良配体,通常在Pd催化的C-H官能化中是无效的导向基团。在之前的研究中,Sanford[10]利用肟醚(3)为有效的引导基团用于PhI(OAc)2/Pd(OAc)2催化氧化的sp2和sp 3 C-H乙酰氧基化反應,然而,从官能化产物(4)中除去肟醚保护基是比较困难的(图2)。
因此,Sanford试图在C-H官能化时用乙酰基修饰肟基,使其转化为具有更多配位点的O-乙酰肟醚衍生物,然后在碱性环境下脱去该修饰基团得到相应的酮产物。实验证明,原位产生的O-乙酰肟(6)作为Pd(II)催化C-H官能化中的导向基团在催化条件下稳定存在,且可在一定条件下脱去乙酰保护基,破坏肟醚骨架得到酮(7)。而端位的sp3 C-H键经乙酰氧基化构建C-OAc(6),后经脱乙酰基作用生成了醇(7),间接实现了C-H键的羟基化(图3)。Sanford尝试了14个例子,涉及脂肪族肟、芳香族肟以及环烷基肟等,在钯催化氧化C-H键活化诱导中以33-86%的产率得到O-乙酰肟衍生物。随后,得到的O-乙酰肟衍生物通过各种方法(K2CO3/NaHSO3,K2CO3/NaHSO3/H2,K2CO3/p- TsOH/ZnCl2等)除去乙酰修饰基团得到相应的醇、胺类、酮和含氮杂环。
2012年,Ren 和Dong[11]通过肟醚诱导和钯催化氧化作用活化sp3 C-H键,生成乙酰氧基取代的肟醚衍生物(10),随后肟醚10经两种不同氧化途径得到化学结构不同的1,2-二醇衍生物(11和12)。他们指出脂肪族sp3 C-H键进行选择性官能化,从一元醇衍生物经过碳氢活化再氧化得到两种化学结构不同的1,2-二醇衍生物(图 4)。其中,肟用作该转化的外导向基团(DG)和醇替代物。由此可知,在C-H活化中使用外导向基团(exo-DG)可能会发现新的转化和易消除的诱导基团。值得一提的是,活化产物经脱乙酰基后得到相应的醇(12),与Sanford除去乙酰修饰基团的研究有着异曲同工之妙,但是Ren 和Dong采用的条件更加温和,保护了肟基团骨架,只是选择性地脱去乙酰基团。同样地,该反应可认为是C-H键的间接羟基化。
Ren 和Dong对sp3 C-H键活化的反应条件进行了优化和探索,以肟衍生的2-丁醇(A:2,6-二甲基苯基,B:H,8)为底物,发现Pd(OAc)2 (10 mol%)/PhI(OAc)2/AcOH/Ac2O体系是构建C-OAc键产率最高的反应条件。在此基础上,他们对该官能化反应进行了底物适用性范围探索(12个例子),发现均可以与肟醚的sp3 C-H键发生选择性氧化(44%~%)。
除了-CH3的C-H键可以活化之外,Ren 和Dong发现环状肟醚中的亚甲基(-CH2-)和桥头位上的次甲基(-CH)同样可以被官能化。另外,薄荷醇衍生的肟醚在进行钯催化氧化时发生了氧化骨架重排。
关于C-H活化产物中残留诱导基团的消除问题,Johnson[12]在合成甾体生物碱Paspaline的研究中也进行了探讨。与Sanford的O-乙酰肟醚经诱导基团转移生成酮醇的研究类似,Johnson利用HCl和DMP使O-Bn和O-Ac键断裂,除去导向基团,生成相应的醛酮(图5)。Johnson将Sanford报道的C-H键乙酰氧基化条件直接用于肟(13),以79%的产率得到了单一非对映异构体的单乙酸酯(14),该转化完成了最终季铵中心的组装,随后经脱保护和氧化作用生成酮醛化合物(15)。
1.2 ?C-H键的酰胺化
2006年,Che[13]报道了钯催化级联活化的sp2 C-H键和sp3 C-H键的分子间酰胺化反应,研究并开发了一种基于级联螯合-定向环化钯催化C-H键的酰胺化方案。该方案可实现未激活的sp2和sp3 C-H键的活化催化并构建C-N键,具有显著的区域和化学选择性。
Che发现以O-甲基肟为诱导基团的钯催化C-H键活化可以得到极高转化率和良好收率的邻位酰胺化产物(图6)。由对位取代的苯甲醛衍生的O-甲基肟有效地转化为相应的邻位C-H酰胺化产物,且具有良好的区域选择性。
1.3 ?C-H键的芳基化
2008年,Yu和Shi[14]等人实现了以钯催化、肟醚基团导向的O-甲基-(E)-2甲基苯甲醛肟与芳基硼酸之间的交叉偶联反应,构建C-Ar键。以Cu(OTf)2及苯醌(BQ)为氧化剂,通过2,6-二甲氧基吡啶(DMOP)抑制芳基硼酸与C=N键的直接加成,从而实现邻位C-H键的活化,构建C-Ar键(图7)。Shi还进行了取代硼酸的底物耐受性探索,发现各类电性的芳基硼酸均可以得到较高产率的目标化合物,且具有良好的官能团耐受性。值得一提的是,对于空间位阻较大的芳基酮肟,在相同的条件下,其活化转化率较低,然而在当体系中去除2,6-二甲氧基吡啶时,则可以得到良好的转化率。
1.4 ?C-H键的酰化
2010年,Chan 和Yu[15]开发了一种新型的钯催化C-H键活化方案,以叔丁基过氧化氢(TBHP)作为氧化剂使芳基酮肟和醛发生交叉偶联,进行直接C-H键酰化。该方法将O-甲基肟作为C-H活化的导向基团,实现了与醛的偶联并且具有显著的区域选择性。Chan 和Yu 针对该方案进行了条件优化,最终确定Pd(OAc)2 (5 mol%)/TBHP/AcOH/toluene/ 100 体系为该反应的最优条件,随后他们进行了底物适用性范围探索,发现该酰化反应表现出优异的官能团耐受性,并且脂肪族和杂芳族醛都可以有效地偶联到肟上(图8)。
1.5 ?C-H键的硝基化
2013年,Xu和Zhang[16]成功实现了钯催化的螯合辅助C-H硝化的硝基芳烃的区域特异性合成。该硝化可不受取向规则的影响,使用O-甲基肟基作为可去除的导向基团,芳基酮(23)经由Pd-催化的C-H-键的邻位硝化的三步过程实现各种邻硝基芳基酮(26)的区域特异性。Xu和Zhang针对C-H活化构建C-N键进行了一系列的底物适用性范围探索。结果显示,各官能团均有较好的耐受性。同时,他们也进行了去除诱导基团的官能团耐受性范围测试,所得数据同样表现出良好的收率。此外,Xu和Zhang开展了一系列氮杂芳烃如2-芳基喹喔啉、吡啶、喹啉和吡唑的C-H鍵硝化,且具有极好的化学和区域选择性(图9)。
作者认为该硝化进程涉及在氧化条件下,Pd(II / III)和/或Pd(II / IV)参与催化循环的银介导的自由基机制。目前该方案也已成功地应用于芳基酮经过区域特异性合成邻硝基芳基酮,包括诱导基团引入、C-H键的邻位硝化和诱导基团去除。目前对C-H键进行硝基化的策略具有两个特点:(1)在定向规则的背景下不受定位规则的影响,特异性硝化C-H键,并具有优异的硝基化选择性;(2)适用的官能团范围广泛,在中性条件下的底物耐受性良好,且不需要预先官能化C-H键。
1.6 ?C-H键的羟基化
2015年,Jiao 和Liang[17]报道了关于配体促进钯催化的肟醚诱导芳烃的邻C-H羟基化的研究。
他们开发出一种在中性条件下有效的Pd(II)催化肟醚芳烃的邻位C-H羟基化的有效方案,以高效率获得具有高度选择性的单羟基化产物。利用Oxone(过硫酸氢钾复合盐,一种廉价易得且安全的试剂,经常用作终端氧化剂和氧气来源)作为催化剂,在配体存在下进行C-H活化,配体的主要作用在于可加速C-O键还原消除进程。换言之,促进Pd(0)/ Pd(II)的催化循环。Jiao和Liang 以芳基肟醚为活化底物进行了反应条件的优化,发现Pd(OAc)2 (5 mol%)/PPh3 或 DEAD (10 mol%)/TCE体系为获得高产率C-H羟基化产物的最佳条件。随后,他们探索了底物适用性范围,实验数据表明该方法具有丰富的底物耐受范围,对具有缺电子基团的底物同样适用(图10)。此外,该C-H羟基化已经成功应用于某些药物的结构修饰。
图10 ?芳香肟的邻位C-H羟基化
1.7 ?不对称烯烃双氧化
2013年,Sanford[18]报道了Pd催化的不对称手性配体导向的烯烃1,2-双氧化反应。Sanford通过将手性肟醚引导基团束缚在烯烃底物上以控制反应的非对映选择性。作为导向基团,手性肟醚可用于控制高价钯催化的烯烃双氧化的立体化学选择性。
Sanford在选择手性肟醚为诱导基團进行不对称烯烃双氧化时,主要依据三个关键点:
(1)已知肟醚可有效地诱导其他钯催化反应(尤其是C-H官能化);
(2)底物可以由烯丙醇和手性酮制备获得且工艺简单易实行;
(3)各种手性非外消旋酮类化合物可以很快获得。Sanford尝试13个不同的手性肟醚进行不对称烯烃双官能化,涉及环己基肟醚端烯、桥环肟醚端烯类以及2-取代环己基肟醚端烯的C-H活化,均实现了不对称烯烃的双活化,且dr值最高为9∶1(图11)。
此外,Sanford还研究了反式和顺式内烯烃的非对映选择性双苯甲酰化反应,同样得到了理想的dr值,关于这种转化的机理,有文献报道称该转化通过肟醚配位,氧化Pd(II)为Pd(IV),然后以C-O键的形式进行还原消除。然而作者认为,在此背景下,有几个尚未解决的细节对反应的立体化学结果至关重要。 特别是最初的步骤可以通过反式或顺式氧化还原来进行[19,20],而且最终的还原消除可以通过直接消除或SN2型机制进行[19-21]。在非手性系统中,这四条途径将产生两种不同的产物(anti 和 syn)。
1.8 ?C-H键的环化反应
环醚类广泛存在于某些生物大分子中,尽管目前报道了许多由醇合成环醚的有效方法(例如各种SN2反应),但最具潜力的是由O-H键和未活化的sp3C-H键之间的脱氢偶联合成环醚的方法,它可避免待环化位置的预活化且反应效率高(图12)。
Dong[22]在2015年报道了用内醇亲核试剂对未官能化的sp3 C-H键进行钯催化官能化合成环醚的方法。由肟修饰的醇为导向基团,选择性地在醇的β位发生环化反应,从而合成一系列四至七元环的脂肪族环状醚。Dong以单保护二醇(2,6-二甲氧基苯甲醛肟n=1)作为初始底物进行条件优化,发现用10 mol%Pd(OAc)2和1.3当量的PhI(OAc)2在HOAc中,反应温度为100 ℃下以64%的收率形成四氢呋喃产物(图13)。继而Dong研究了该方法的底物官能团耐受性,得出该反应具有丰富的底物适用性范围,无论是连接的伯,仲和叔游离羟基都可以得到相应的环化产物。另外,苄基和甲硅烷基保护的醇也可以直接偶联。
关于该环化反应机制,作者提出了sp3 C-H活化/分子内SN2途径。首先,经过定向的C-H钯催化,在钯被氧化成高氧化态(PdIV)后,可发生分子内SN2反应,产生氧鎓中间体,同时将钯(Pd IV)还原成钯(PdII)。随后,乙酸根离子经过去质子化或去苄基化后得到相应的环醚产物。
Jiang 和Zhu[23]在2016年报道了钯(II)催化的肟与乙烯基叠氮化物的交叉偶联合成3-取代的和1,3-二取代的异喹啉。肟作为转化中的导向基团和内部氧化剂,不需要化学计量的外部氧化剂。使用简单的原材料,不需要添加剂,无毒副产品,操作简单,且反应条件温和,为合成不同种类的异喹啉提供了有效方案。
肟34作为诱导基团和内部氧化剂经过两种途径([4+2]环化和[3+3]环化)生成取代异喹啉,Jiang 和Zhu根据这两种反应类型做了条件优化,发现在Pd(OAc)2/toluene/90 ℃体系下,可以以良好的收率合成异喹啉35,且在最优条件下进行了底物官能团耐受性探索,实验显示该反应表现出良好的底物耐受性。再者,体系Pd(OAc)2/CH3CN/130 ℃为[4+2]环化反应提供了良好的条件,以良好的收率合成异喹啉33,且具有丰富的底物适用范围(图14)。
1.9 ?C-H键的卤化反应
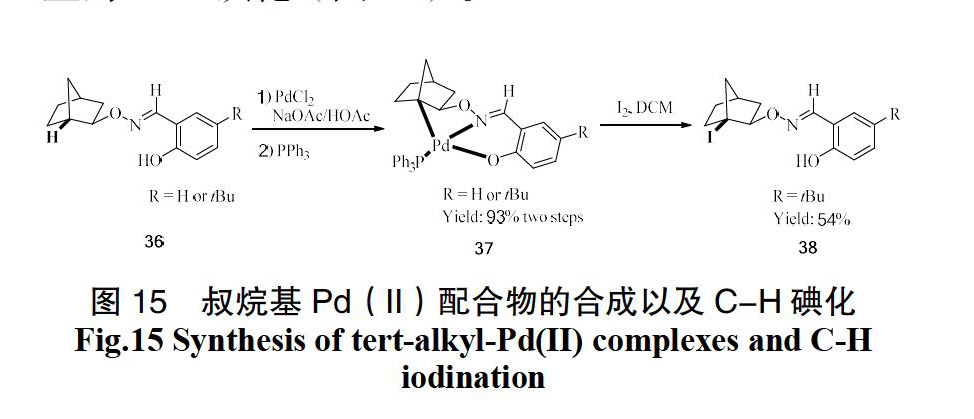
2016年,Dong和Ren[24]制备并表征了通过C-H活化的叔烷基Pd(II)配合物的第一个实例,由肟的外导向作用,在桥环顶端位置顺利发生环钯化生成叔烷基Pd(II)配合物。这项研究表明,环钯化反应可以发生在叔碳位的sp3 C-H键,为钯催化的次甲基C-H键官能化提供了重要的机理信息。此外,Dong还用碘处理钯(II)配合物导致次甲基碳上的C-H碘化(图15)。
2 ?总结和展望
肟醚类化合物,在医药领域可以作为一类高效抗菌消炎剂,另外有研究表明,肟醚具有抗肿瘤、抗惊厥等生物活性,是某些具有生物活性药物的关键结构单元。在植物学领域,是一类抗植物病毒的特效药。在有机合成领域,是一类具有多种用途的前体和中间体,可通过多种有机转化生成一系列含氮化合物。在有机催化领域,尤其是C-H键的催化活化,是一种应用广泛的诱导基团,可参与诱导C-H键的氧化、羟基化、酰基化、硝基化、卤化、芳基化以及环化等各种活化模式。
目前,有关肟醚诱导的过渡金属催化C-H活化的研究已日趋成熟,然而相较于其他诱导基团,肟醚类作为诱导基团的应用还具有很大的研究空间。随着催化领域的发展,相信会发掘出更多的C-H活化模式,而肟醚将会以更多的形式参与其中。
参考文献:
[1]Babazadeh-Qazijahani M, Badali H, Irannejad H, Afsarian M H, Emami S.Imidazolylchromanones containing non-benzylic oxime ethers: Synthesis and molecular modeling study of new azole antifungals selective against Cryptococcus gattii [J]. Eur J Med Chem, 2014, 76: 264-273.
[2]Parthiban P, Kabilan S, Ramkumar V, Jeong Y T. Stereocontrolled facile synthesis and antimicrobial activity of oximes and oxime ethers of diversely substituted bispidines [J]. Bioorg Med Chem Lett, 2010, 20: 6452-6458.
[3]Chern J-H, Lee C-C, Chang C-S, Lee Y-C, Tai C-L, Lin Y -T, Shia K-S, Lee C-Y, Shih S-R. Synthesis and antienteroviral activity of a series of novel, oxime ether-containing pyridyl imidazolidinones [J]. Bioorg Med Chem Lett, 2004, 14: 5051-5056.
[4]Bernardi L, Cere V, Femoni C, Pollicino S, Ricci A. Organometallic Reactions in Aqueous Media:Indium-Promoted Additions to 2-Pyridyl and Glyoxylic Acid Oxime Ethers [J]. J Org Chem, 2003, 68: 3348-3351.
[5]Sharkova L, Aksanova L, Kucherova N, Zagorevskii V. Ring closure of some O-aryl ethers of ketoximes to benzofurans. Chem. Heterocycl [J]. Compd, 1971, 7: 1482-1486.
[6]Noverges B, Mollar C, Medio-Sim?n M, Asensio G. Palladium- Catalyzed Suzuki-Miyaura Cross-Coupling of α-Halomethyl Oxime Ethers and Site-Selective Cross-Coupling of Dihalo Derivatives [J]. Adv Synth Catal, 2013, 355: 2327-2342.
[7]Fujiwara Y, Noritani I, Danno S, Asano R, Teranishi S. Aromatic substitution of olefins. VI. Arylation of olefins with palladium(II) acetate [J]. J Am Chem Soc, 1969, 91: 7166-7169.
[8]Desai L V, Hull K L, Sanford M S. Palladium-Catalyzed Oxygenation of Unactivated sp3 C-H Bonds [J]. J Am Chem Soc, 2004, 126: 9542-9543.
[9]Neufeldt S R, Sanford M S. O-Acetyl Oximes as Transformable Directing Groups for Pd-Catalyzed C-H Bond Functionalization [J]. Org Lett, 2010, 12: 3532-3535.
[10]Desai L V, Stowers K J, Sanford M S. Insights into Directing Group Ability in Palladium-Catalyzed C?H Bond Functionalization [J]. J Am Chem Soc, 2008, 130: 13285-13293.
[11]Ren Z, Mo F, Dong G. Catalytic Functionalization of Unactivated sp3 C?H Bonds via exo Directing Groups: Synthesis of Chemically Differentiated 1,2-Diols [J]. J Am Chem Soc, 2012, 134: 16991?16994.
[12]Sharpe R J, Johnson J S. A Global and Local Desymmetrization Approach to the Synthesis of Steroidal Alkaloids: Stereocontrolled Total Synthesis of Paspaline [J]. J Am Chem Soc, 2015, 137: 4968?4971.
[13]Thu H-Y, Yu W-Y, Che C-M. Intermolecular Amidation of Unactivated sp2 and sp3 C-H Bonds via Palladium-Catalyzed Cascade C-H Activation/Nitrene Insertion [J]. J Am Chem Soc, 2006, 128: 9048-9049.
[14]Shi B-F, Maugel N, Zhang Y-H, Yu J-Q. PdII-Catalyzed Enantioselective Activation of C(sp2)-H and C(sp3)-H Bonds Using Monoprotected Amino Acids as Chiral Ligands[J]. Angew Chem Int. Ed, 2008, 47: 4882-4886.
[15]Chan C-W, Zhou Z, Chan A S C, Yu W-Y. Pd-Catalyzed Ortho-C-H Acylation/Cross Coupling of Aryl Ketone O-Methyl Oximes with Aldehydes Using tert-Butyl Hydroperoxide as Oxidant [J]. Org Lett, 2010, 12: 3926-3929.
[16]Zhang W, Lou S, Liu Y, Xu Z. Palladium-Catalyzed Chelation-Assisted Aromatic C?H Nitration: Regiospecific Synthesis of Nitroarenes Free from the Effect of the Orientation Rules [J]. J Org Chem, 2013, 78: 5932?5948.
[17]Liang Y-F, Wang X, Yuan Y, Liang Y, Li X, Jiao N. Ligand-Promoted Pd-Catalyzed Oxime Ether Directed C?H Hydroxylation of Arenes [J]. ACS Catal, 2015, 5: 6148?6152.
[18]Neufeldt S R, Sanford M S. Asymmetric Chiral Ligand-Directed Alkene Dioxygenation [J]. Org Lett, 2013, 15: 46-49.
[19]McDonald R I, Liu G, Stahl S S. Palladium(II)-Catalyzed Alkene Functionalization via Nucleopalladation: Stereochemical Pathways and Enantioselective Catalytic Applications[J]. Chem Rev, 2011, 111: 2981-3019.
[20]Park C P, Lee J H, Yoo K S, Jung K W. Efficient Diacetoxylation of Alkenes via Pd(II)/Pd(IV) Process with Peracetic Acid and Acetic Anhydride[J]. Org Lett, 2010, 12: 2450-2452.
[21]Desai L V, Sanford M S. Construction of Tetrahydrofurans by PdII/PdIV-Catalyzed Aminooxygenation of Alkenes[J]. Angew Chem Int Ed, 2007, 46: 5737-5740.
[22]Thompson S J, Thach D Q, Dong G. Cyclic Ether Synthesis via Palladium-Catalyzed Directed Dehydrogenative Annulation at Unactivated Terminal Positions[J]. J Am Chem Soc, 2015, 137: 11586?11589.
[23]Zhu Z, Tang X, Li X, Wu W, Deng G, Jiang H. Palladium-Catalyzed C-H Functionalization of Aromatic Oximes: A Strategy for the Synthesis of Isoquinolines[J]. J Org Chem, 2016, 81: 1401-1409.
[24]Ren Z, Dong G. Direct Observation of C?H Cyclopalladation at Tertiary Positions Enabled by an Exo-Directing Group[J]. Organometallics, 2016, 35: 1057-1059.
